
Recently Dr. Balistreri gave our group an excellent lecture. I have taken some notes and shared some slides. There may be inadvertent omissions and mistakes in my notes.
Key Points:
- Producing enough bile acids and recycling bile acids in enterohepatic circulation is crucial for bile acid flow. In addition, there are ‘good’ bile acids like cholic acid that have trophic properties and ‘bad’ bile acids like lithocholic acid that cause liver toxicity
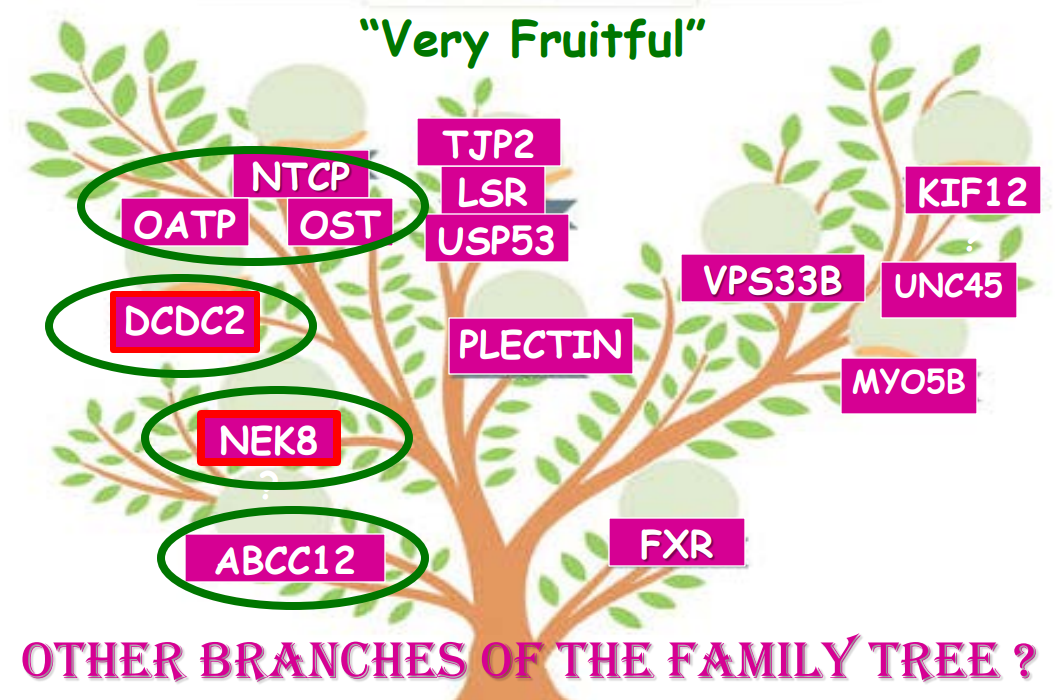
- In addition to defects in the metabolic pathway of bile acids, discoveries identified defects in the membrane transporters (eg. FIC1, BSEP, MDR3), trafficking proteins (eg. MYO5B, VPS33B), nuclear control receptors (eg. FXR), and tight junction proteins (eg. TJP2). Tight junction protein defects are associated with bile leakage from bile canaliculus
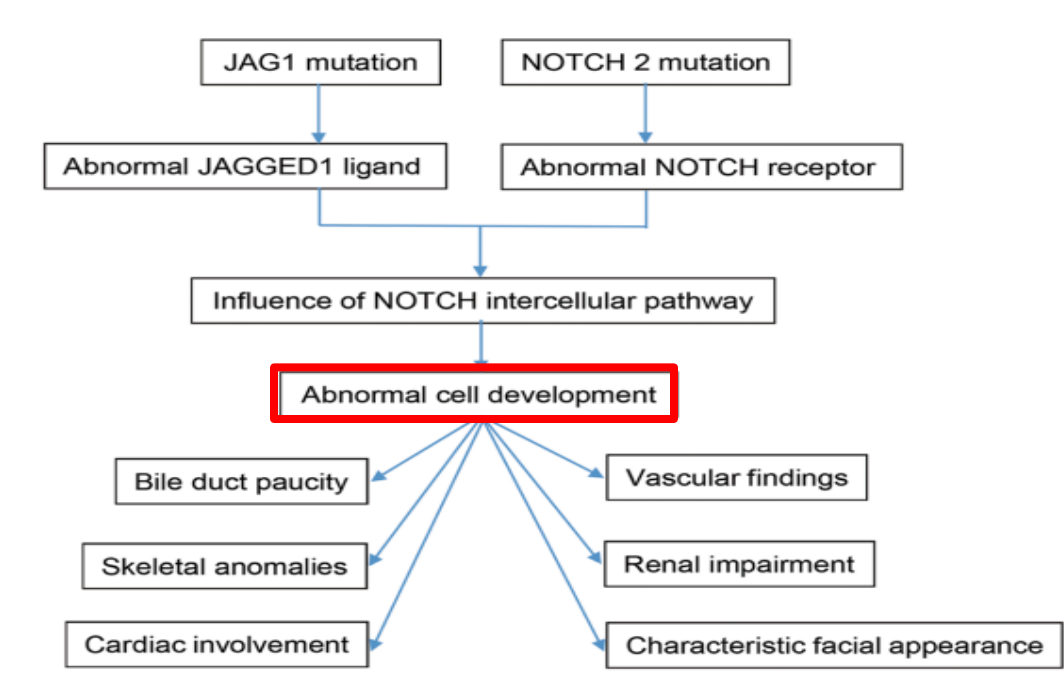
- Alagille syndrome, a disorder of embryogenesis, related to JAG1-NOTCH2 signaling pathways affects organs throughout the body
- Many of these genetic mutations are now being identified in adults with unexplained liver diseases (eg. intrahepatic cholestasis of pregnancy and cryptogenic cirrhosis)
- Cholestasis panels and whole exome sequencing are important tools
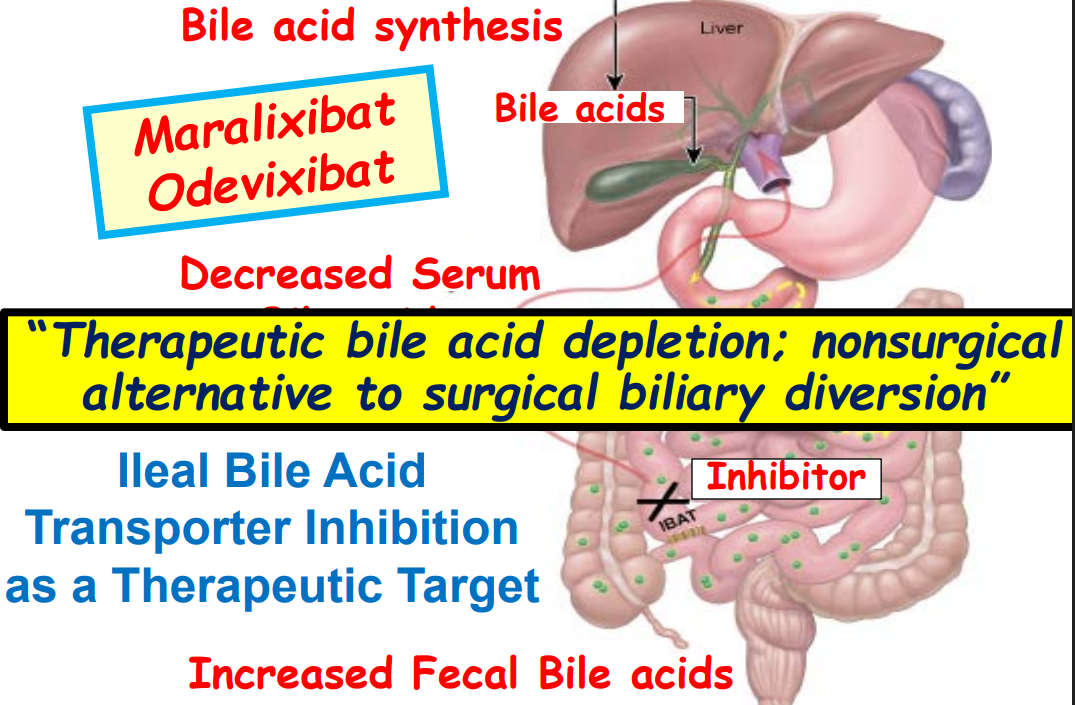
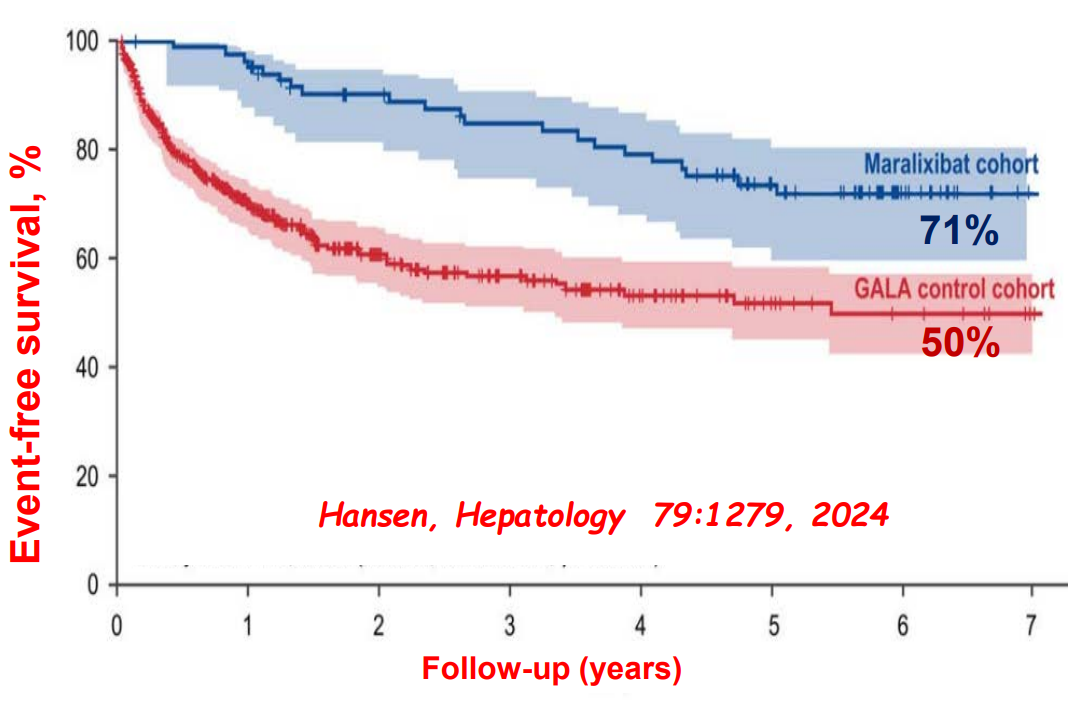
- Ileal bile acid transporter (IBAT) inhibitors have emerged as important therapies for conditions like Alagille which were previously treated with biliary diversion
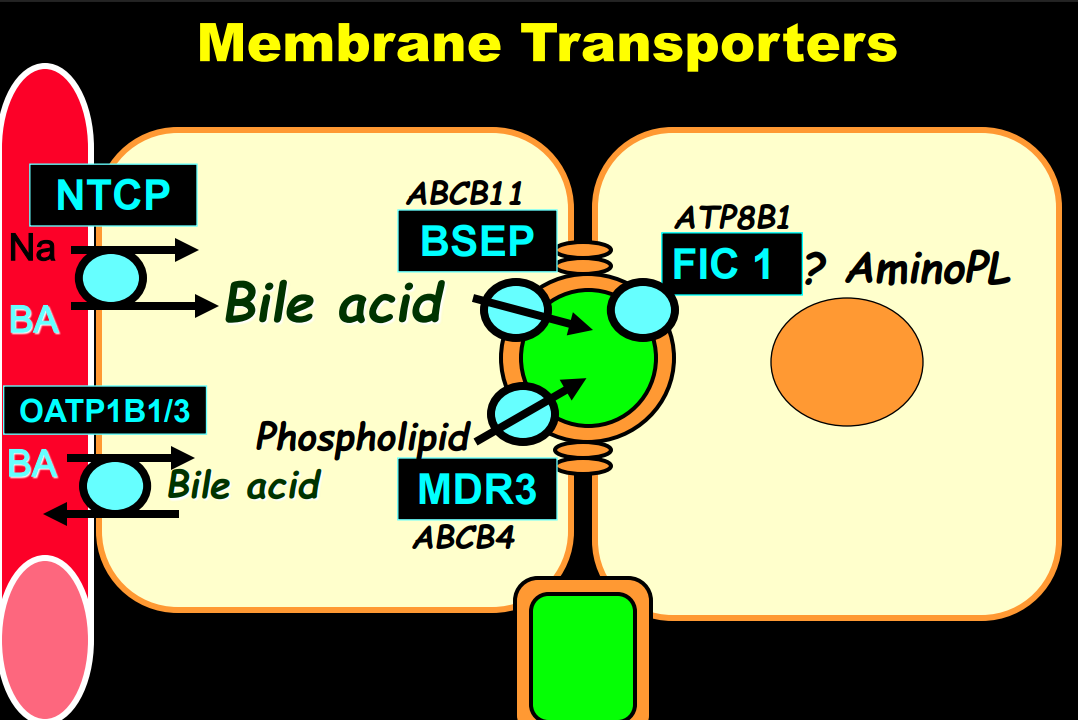




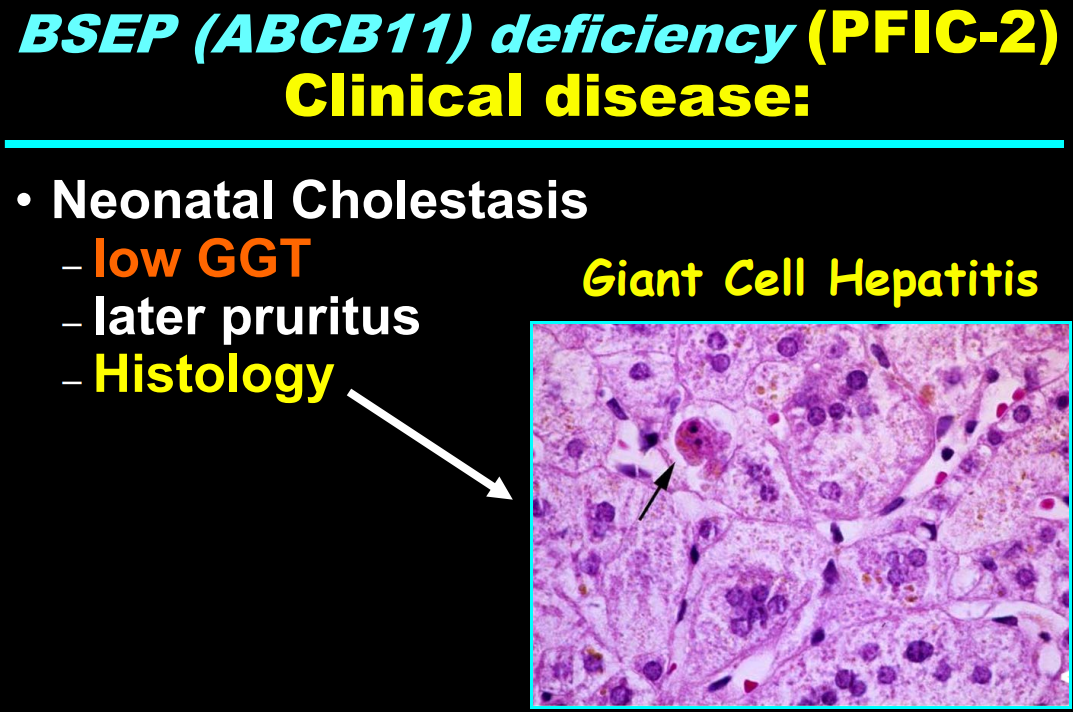


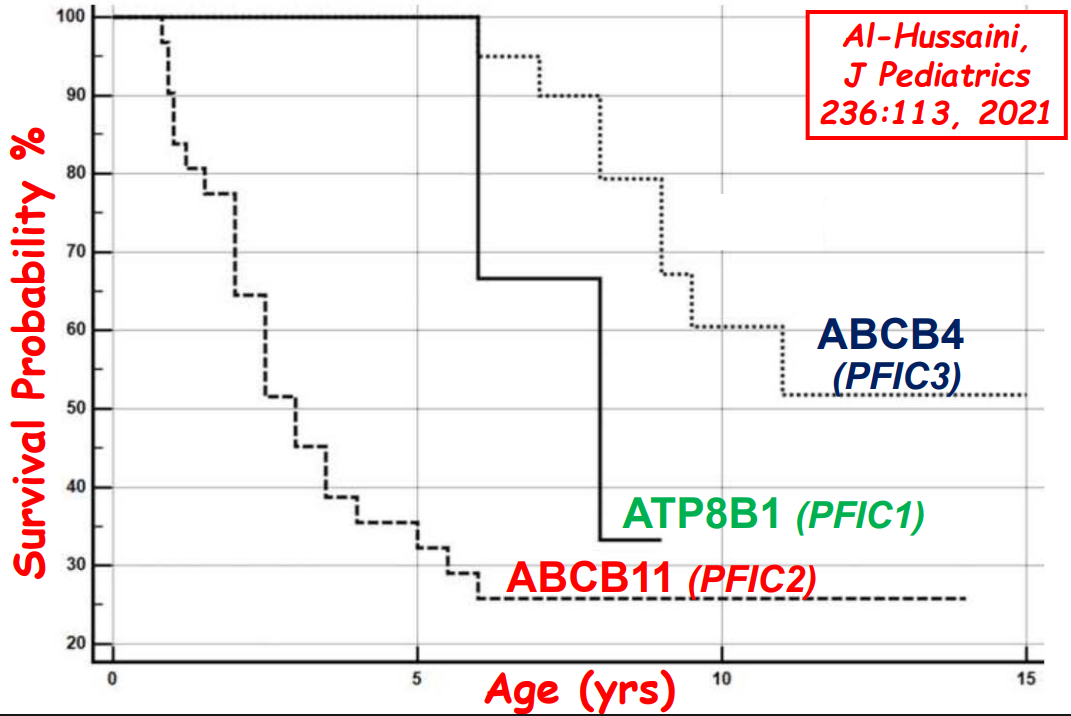


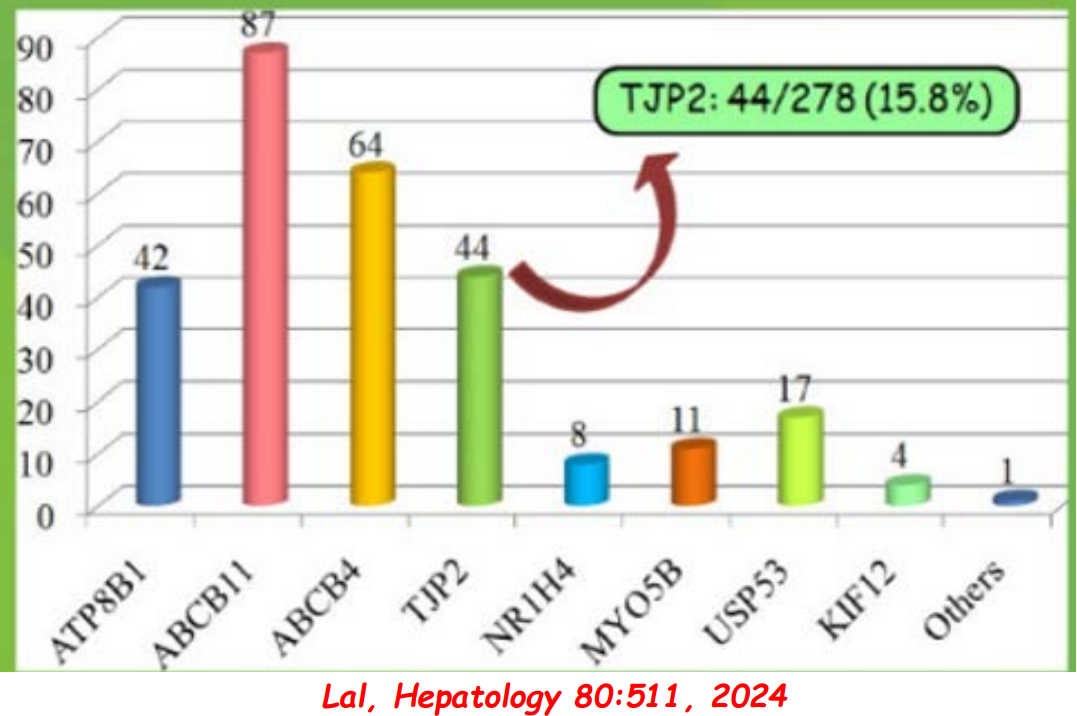




Cholestasis Evaluation:











My take: This lecture really shows how the field of pediatric liver disease has been a puzzle. Now one can see how almost all of the pieces of the puzzle work together.
Related blog posts:
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 1)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 2)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 3)
- Year-in-Review for Pediatric Hepatology (2024)
- Online Aspen Webinar (Part 2) -Abnormal Liver Enzymes in a Tween (2022)
- Medical Progress: Toward Hepatitis C Elimination (2020)
- Immune Mediated Disorders Associated with TNF Inhibitors Can Involve the Liver Too
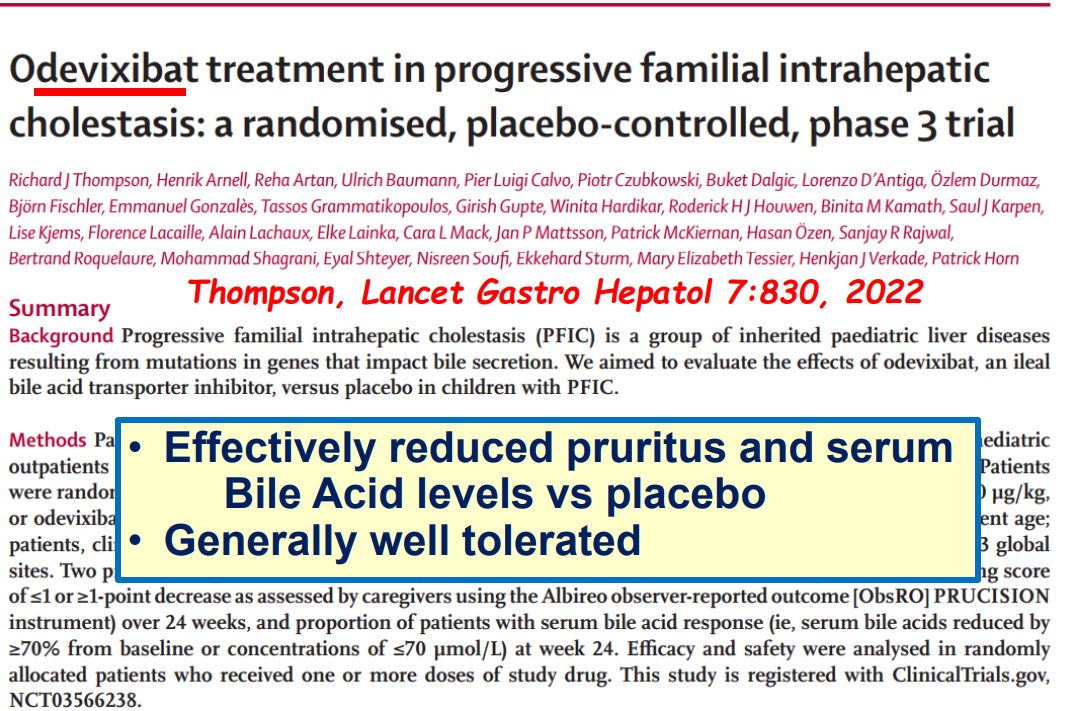
- Efficacy and Safety of Odevixibat with Alagille Syndrome (ASSERT Trial)
- Relooking at 6-Year Data of Maralixibat for Alagille Syndrome
- Six Year Data for IBAT Inhibitor Treatment for Alagille Syndrome
- Lecture: IBAT Inhibitor for Alagille Syndrome
- GALA: Alagille Study
- NASPGHAN Alagille Syndrome Webinar
- Intracranial Hypertension & Papilledema with Alagille Syndrome
- Explaining Differences in Disease Severity for Alagille Syndrome
- Gene Therapy for Alpha-One Antitrypsin Deficiency
- Alpha-1-Antitrypsin Deficiency
- Identifying Biliary Atresia in Infants: New Guidelines
- Updated Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 (MMP-7) for Biliary Atresia
- Why Didn’t Screening for Biliary Atresia Improve Outcome In This Study?

