J Allam et al. NEJM 2024; 390-71-76.A Swell Diagnosis
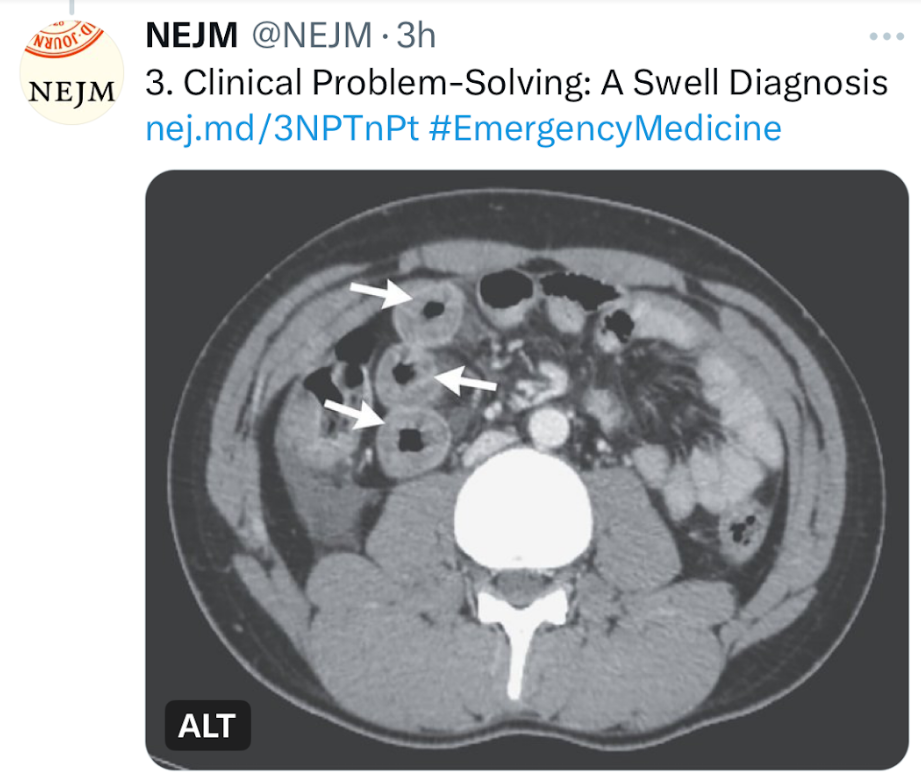
This clinical problem-solving case report describes a previously healthy 19 yo male with sudden-onset severe diffuse abdominal pain. His ED evaluation was unremarkable (including labs [CBC/d, CMP, Lipase, CMP, thryotropin, ESR, CRP and UA], and EKG). He was discharged after 3 hours. Over the next 10 years, he presented to the ED on numerous occasions with the same symptom complex and normal labs/mostly CT scans. One CT scan showed small-bowel thickening thought to be due to AGE. Ultimately, his symptoms increased to every 2 weeks. Extensive evaluations (multiple panendoscopies, MRCP, MRE, U/S, and infectious workup) were undertaken and numerous treatments were given without benefit. Ultimately, a CT scan showed remarkable circumferential wall thickening in the jejunum (see below). This led to evaluation for hereditary angioedema.
The article serves as a good review of this disorder and of the differential diagnosis.
Key points:
- Background: Hereditary angioedema, which is due to a deficiency of functional C1 inhibitor protein, is a rare autosomal dominant genetic disorder that affects approximately 1 in 50,000 persons worldwide.1
- Mean age of onset : 8 to 12 years, and symptoms often worsen during puberty.1
- Presentation: The disease is characterized by recurrent episodes of swelling in various parts of the body and can be severely debilitating. The hallmark symptom of hereditary angioedema is localized swelling of the skin and submucosal tissues (in the face, lips, throat, hands, feet, or genitalia) that is nonpitting, nonpruritic, and not accompanied by urticaria. Triggers include emotional stress, physical trauma, infections, physical exertion, and surgery and other medical procedures.
- Abdominal pain: In a series of 149 patients with hereditary angioedema who had 521 attacks, 49% of the episodes were characterized by isolated abdominal pain.5 Abdominal attacks are generally not associated with fever, peritoneal signs, or leukocytosis.
- Laryngeal edema occurs in approximately 0.9% of all attacks7 and may be life-threatening, leading to asphyxiation and death.
- Differential Diagnosis: Some of the rare diagnosis that were discussed: acute intermittent porphyria, familial Mediterranean fever (FMF), mastocytosis, and eosinophilic gastroenteritis.
- Pathophysiology: Deficiency of C1 inhibitor protein: The majority of cases of hereditary angioedema are caused by either decreased levels (type I) or reduced functionality (type II) of C1 inhibitor. A third subtype (type III) that is associated with different mutations but the same clinical features is characterized by normal quantitative and functional C1 inhibitor levels.2
- The best screening test: measurement of the level of C4 (which is exhausted as a result of uncontrolled activation of the complement pathway when C1 inhibitor is deficient or dysfunctional); results may be normal in 10% of patients between attacks. Quantification of C1 inhibitor levels can then be performed to differentiate between the low levels in hereditary angioedema type I and the normal levels in hereditary angioedema type II.
- Treatment: Food and Drug Administration–approved agents include human plasma–derived C1 inhibitor concentrate, recombinant human C1 inhibitor, icatibant (bradykinin B2 receptor antagonist), and ecallantide (kallikrein inhibitor).9 Each has been shown in randomized, controlled trials to decrease the median time to symptom relief.10-14 The first three therapies may be administered by the patient, whereas ecallantide requires support from a health care professional for management of possible anaphylaxis, which is reported in up to 4% of patients.9 Most treatments are used on-demand, though some patients benefit from long-term prophylaxis.
- Preprocedure prophylaxis: Preprocedural prophylaxis is recommended for patients undergoing procedures that may trigger an attack (e.g., dental surgery, endotracheal intubation, or an endoscopic procedure). C1 inhibitor concentrate can be administered intravenously before the procedure, or treatment with an anabolic androgen (e.g., danazol or stanozolol) can be started 5 days before and continued for 2 to 5 days after the procedure9; …Fresh frozen plasma may be used as a second-line therapy when these therapies are not available
My take: This case “highlights the importance of maintaining a high clinical suspicion for hereditary angioedema in patients with episodic severe abdominal pain and negative workup for other illnesses” even in patients without other manifestations.
Related blog posts:
- Overlooking Important Detail$ in Hereditary Angioedema Treatment
- Intermittent Abdominal Pain and Intestinal Swelling –a Mystery? This case highlights how some medications (eg. lisinopril) can trigger angioedema