Two more studies have shown the effectiveness of CFTR modulators for subsets of patients with cystic fibrosis.
- JL Taylor-Cousar et al. NEJM 2017; 377: 2013-23
- SM Rowe et al. NEJM 2017; 377: 2024-35.
In the Taylor-Cousar study, the authors treated patients with homozygous Phe508del cystic fibrosis with either combination tezacaftor-ivacaftor or placebo for 24 weeks. Combination therapy resulted in FEV1 that was 4% higher along with a 35% lower rate of pulmonary exacerbations than placebo.
In the Rowe study which examined patients some retained CFTR function (which occurs ~5% of CF patients), a prospective trial of tezacaftor-ivacaftor had a greater effect on increasing FEV1 than ivacaftor alone. Ivacaftor monotherapy and tezacaftor-ivacaftor combination therapy were both more effective than placebo.
A related editorial (H Grasemann. pgs: 2085-8) helps provide context to help understand the importance of these studies. His key point:
“Although CFTR modulator therapies have measurable beneficial effects on some aspects of the disease, there is still an unmet need for truly effective new therapies to be developed for all persons with cystic fibrosis. The clinical efficacy of the current combination therapies for patients with cystic fibrosis who have the most common CFTR genotype (Phe508del/Phe508del) is suboptimal and falls within the range of established symptomatic therapies, such as nebulized inhaled hypertonic saline or recombinant human DNAse.”

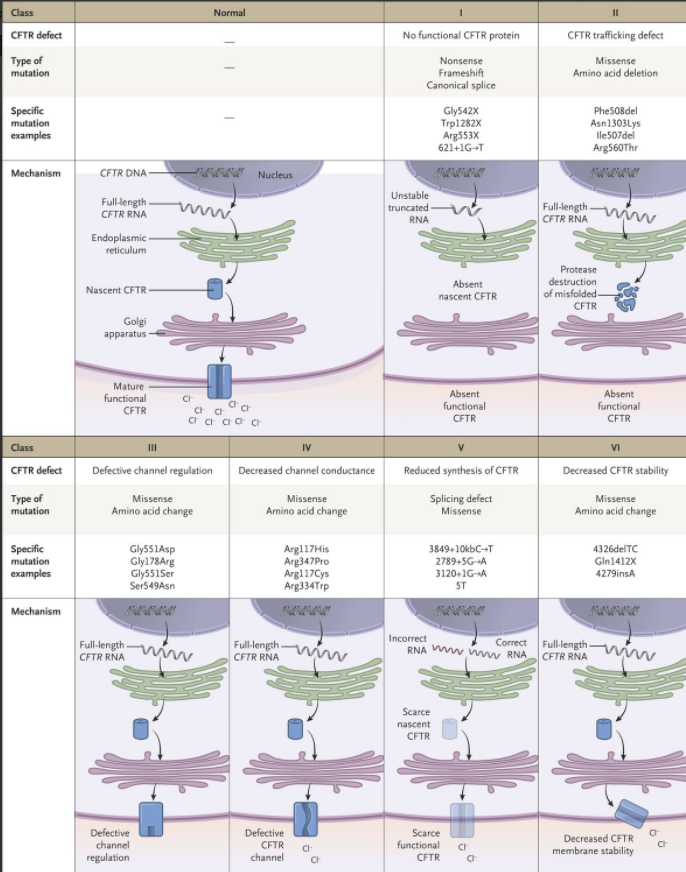
This figure depicts the types of molecular defects: No functional CFTR with framshifts for deletions or insertions (class 1), CFTR trafficking defect due to misfolded protein (class II), defective channel regulation (class III), reduced cholirde conductance (class IV) , reduced synthessis (class V) or decreased CFTR stability (class VI)