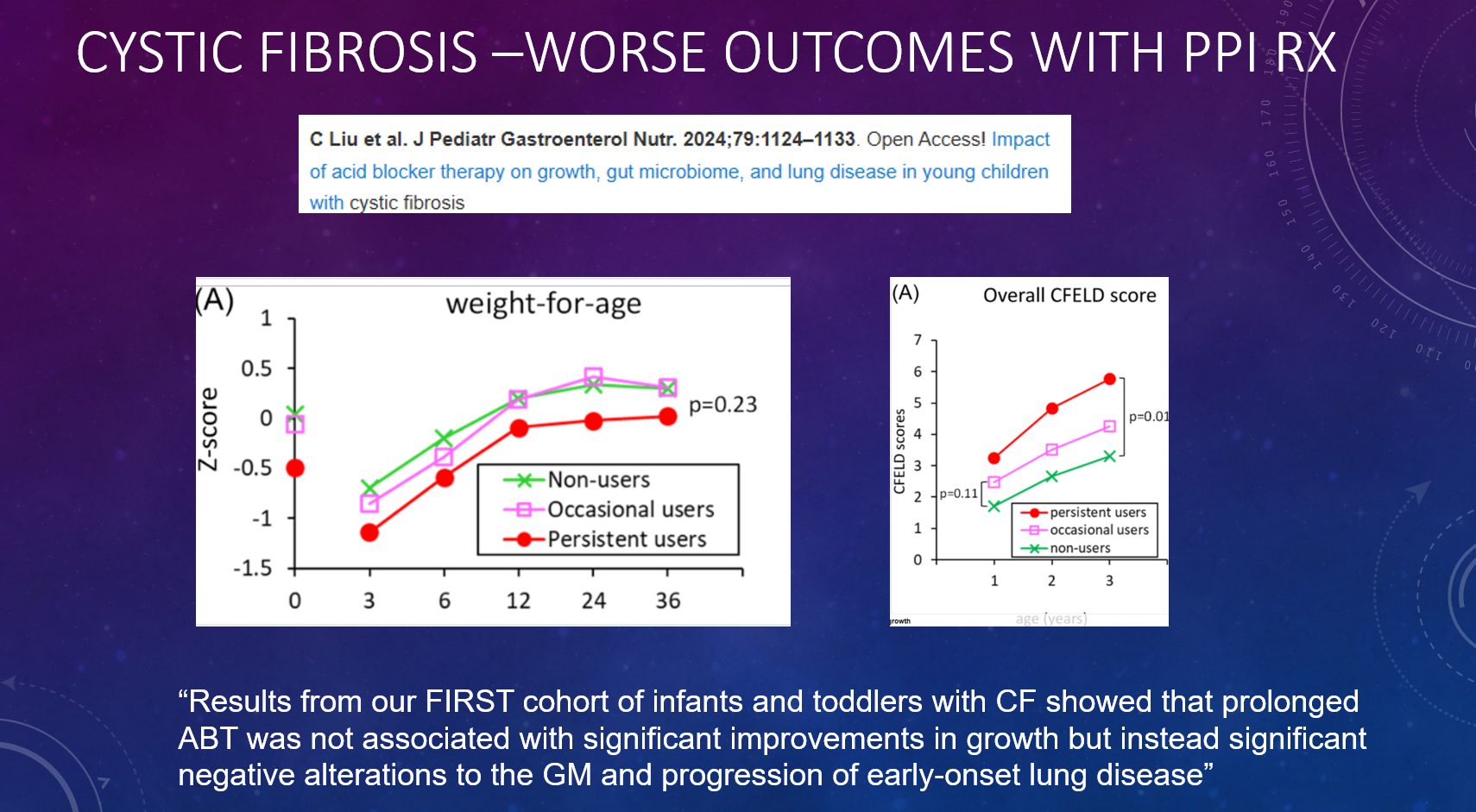
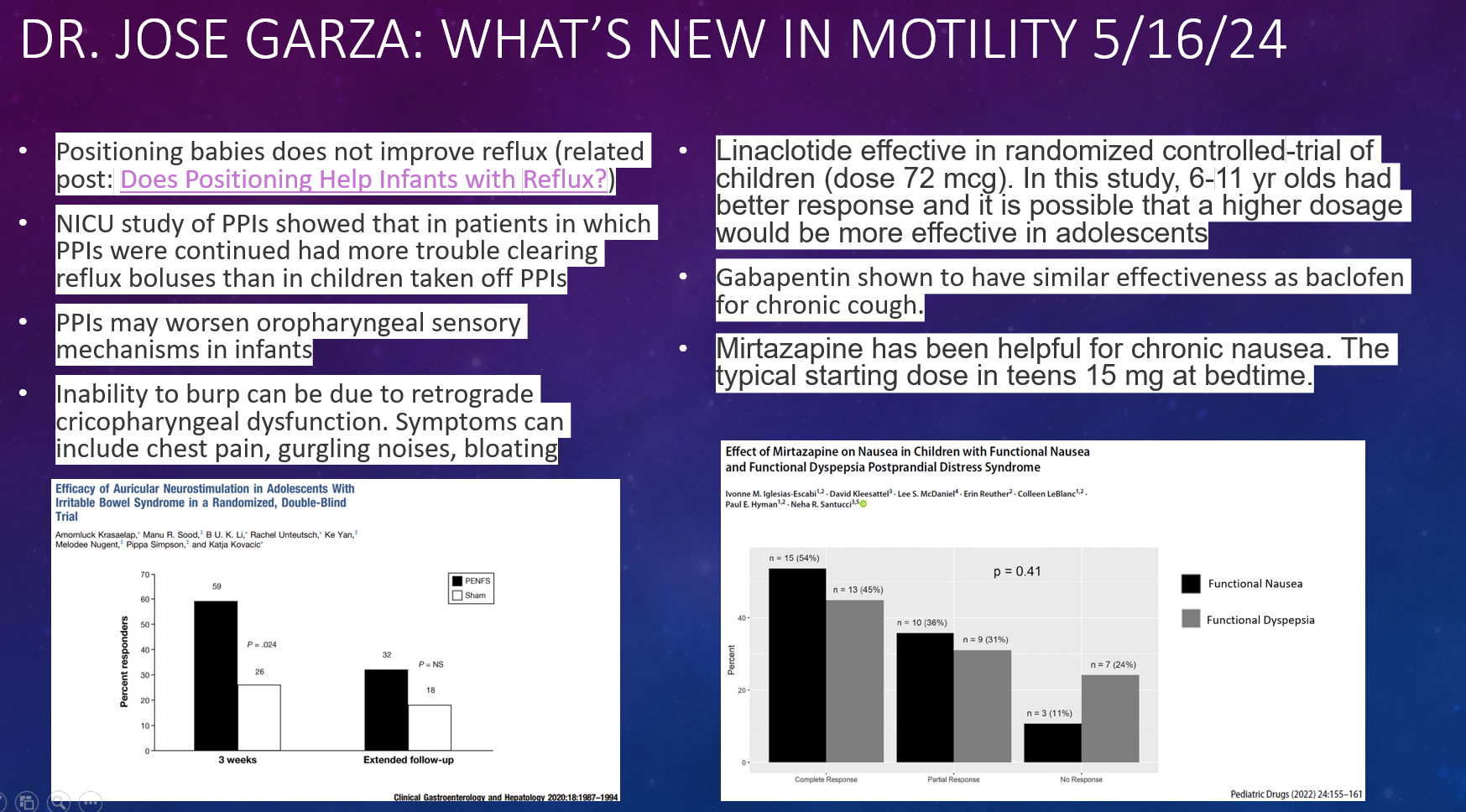
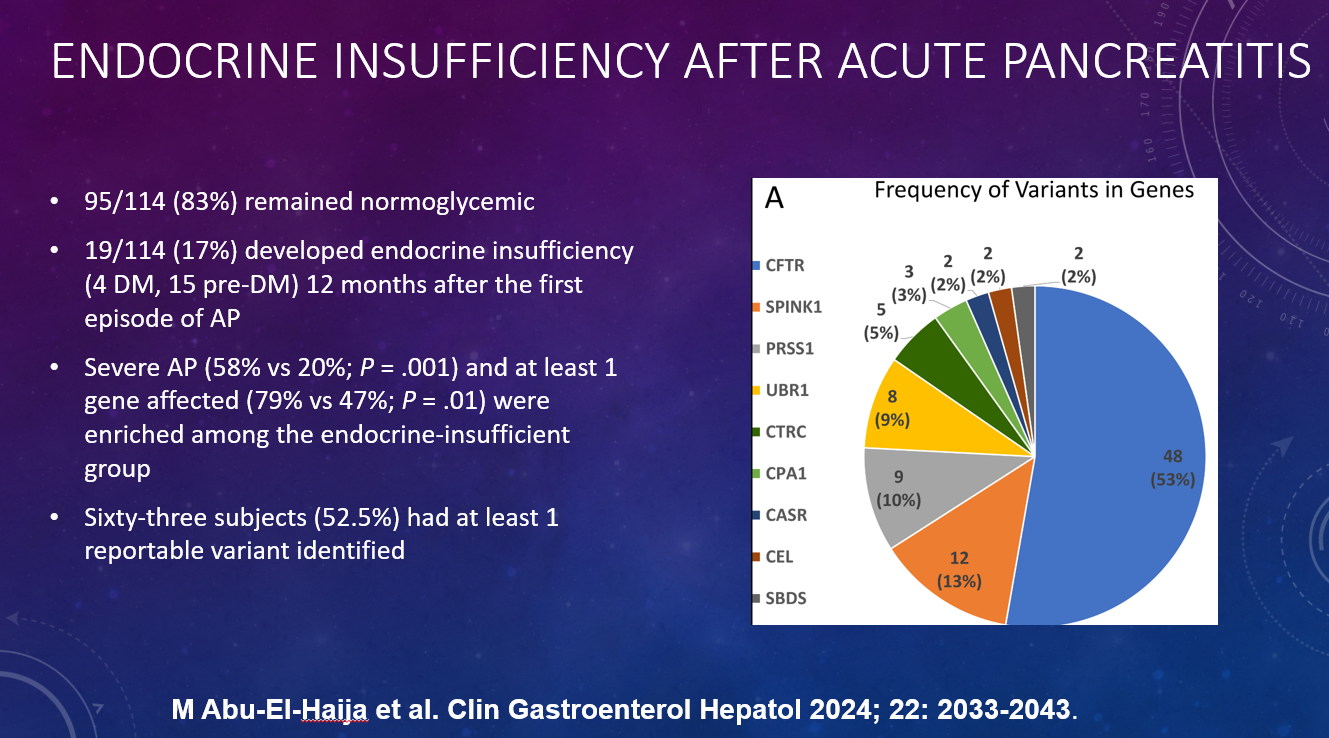
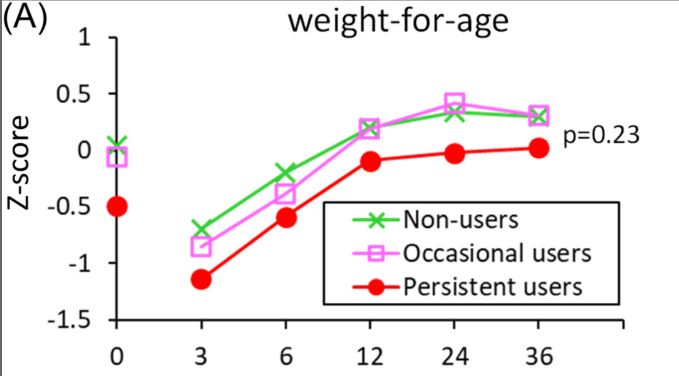
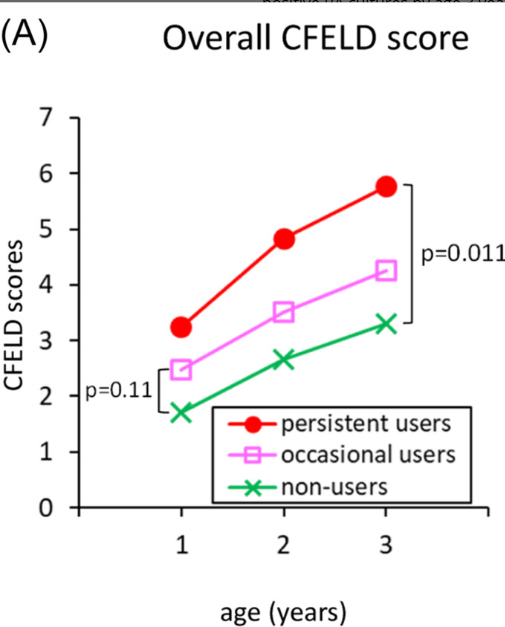
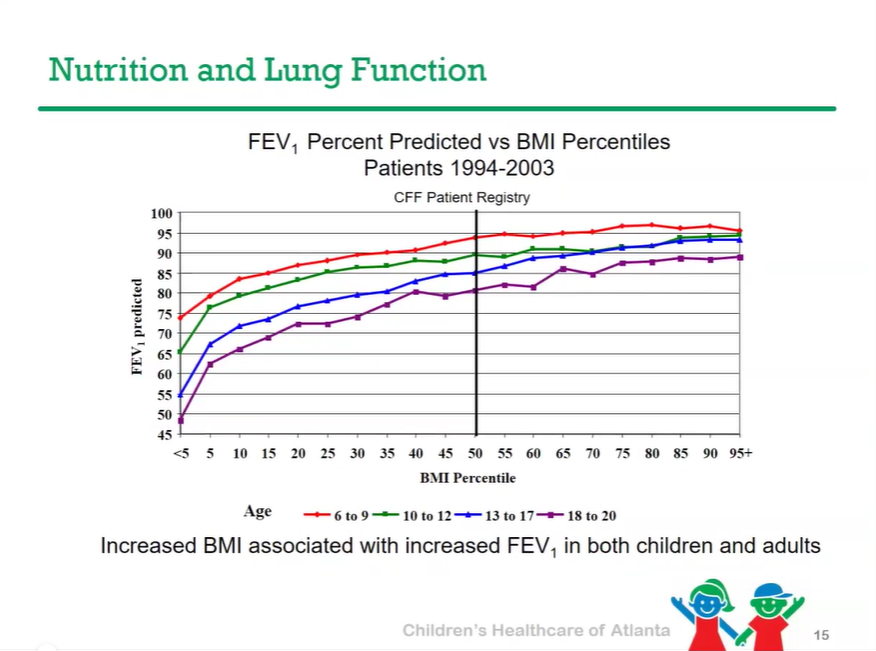
P Saikumar et al. Am J Gastroenterol 2026; 121: 103-111. A Comprehensive Review of Gastrointestinal Manifestations in Cystic Fibrosis in the Era of Highly Effective Modulator Therapy
Thanks to Ben Gold for sharing this article.
Exoocrine Pancreatic Insufficiency (EPI)
- “Mutlicenter studies, KIWI and ARRIVAL, demonstrated increase in FE-1 (fecal elastse-1) after treatment with ivacaftor, signifying improvement in in pancreatic function”
- “The PROMISE study, a large 56-center prospective observational study with participants at least 12 years of age…found ETI (Elexacaftor-tezcaftor-ivacaftor) did not result in improvement in pancreatic function…raises the question if age of initiation of CFTR modular therapy has an influence in recovery of pancreatic function.”
Acute Pancreatitis
- “With the improvement in pancreatic function seen in some patients with CF on HEMT, there have been emerging reports of acute pancreatitis.”
CF Hepatobiliary Involvement
- “Previously referred to as CF liver disease (CFLD), hepatobiliary complications in CF are now recommended to be classified as CF hepatobiliary involvement (CFHBI) or advanced CFLD”
- “Vigilant monitoring of transaminase levels and liver function is crucial after CFTR modulator initiation, because drug-induced liver injury (DILI) is a potential concern with elexacaftor/tezacaftor/ivacaftor (ETI) therapy…A disproportionality analysis of Food and Drug Administration adverse event reporting system data from 2019 to 2022 identified 452 reports of DILI associated with ETI use, demonstrating a statistically significant association”
- “ETI is not recommended for those with Child Pugh Classification C and dose reduction is recommended for Child Pugh classification B in clinically appropriate situation when benefit outweighs the risk”
Nutrition
- “In addition to macronutrient deficiency, Children with Cystic Fibrosis are at risk of deficiencies in fat-soluble vitamins, minerals, and essential fatty acids (38). With an overt focus on nutrition, PERT, and advancements in CFTR modulators, an opposite trend is now being seen, with a higher incidence of obesity and overweight in this population…[and] associated comorbidities such as coronary heart disease, metabolic dysfunction-associated steatotic liver disease, and metabolic syndrome-including hyperlipidemia and diabetes.”
Eating Disorder
- “Disordered eating is relatively a new concern in this population. There are controversial data on the prevalence of eating disorder in PwCF” (people with CF).”
GERD
- “Data indicate that children with CF are 4 times more likely to experience acid gastroesophageal reflux than the general population (59)…RECOVER, a multicenter study of ETI in PwCF displayed decreased GI symptoms, including GERD, after 12 months of treatment (61). Similarly, treatment with IVA was associated with symptomatic relief of GERD at 52 weeks and also showed a decline in extraesophageal reflux in PwCF treated with
- IVA for 12 months (62).”
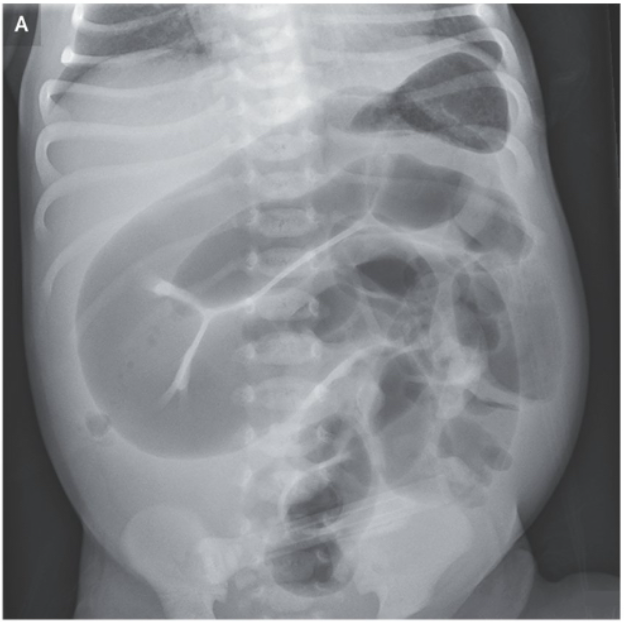
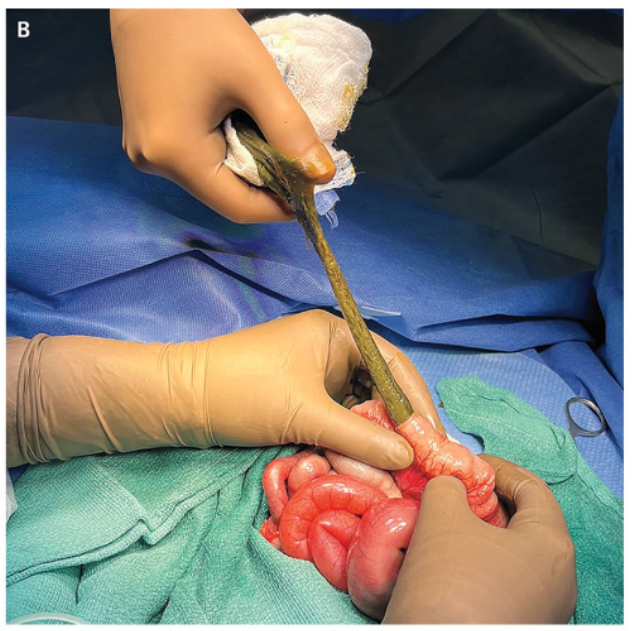
Meconium Ileus
- Recent case reports, “although differing in ETI initiation timing, outcome severity, suggest that prenatal ETI exposure may positively influence MI (69).”
Constipation
- “There are limited data regarding the effect of ETI on constipation, asthe PROMISE trialshowed only small improvements in Patient Assessment of Constipation-Symptoms score and Patient Assessment of Constipation-Quality of Life”
DIOS
- “There is paucity of data describing impact of HEMT [highly effective modulator therapry] on DIOS. By correcting the CFTR dysfunction, HEMT may help reset the ion/fluid balance which is considered one of the main factors in pathogenesis of DIOS.”
My take: “CFTR modulators have revolutionized the management of CF by dramatically altering the course of the disease and improving patient outcomes.” They are also altering the clinical GI manifestations.
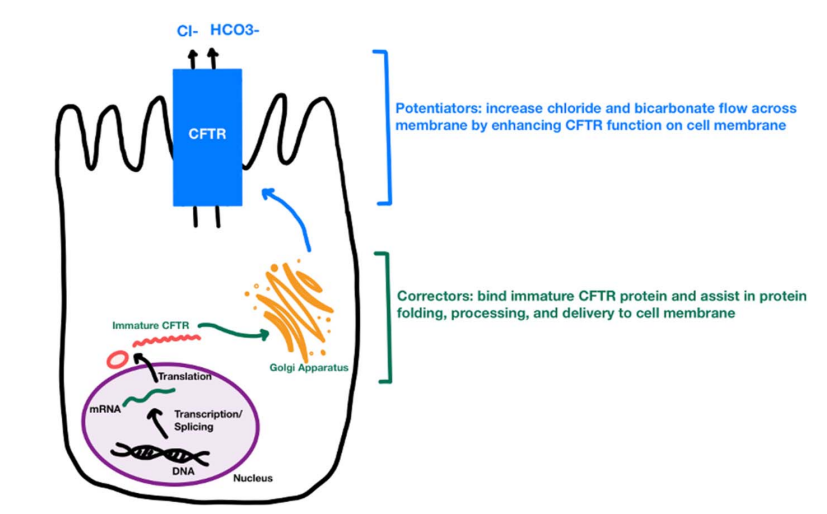
Related blog posts:
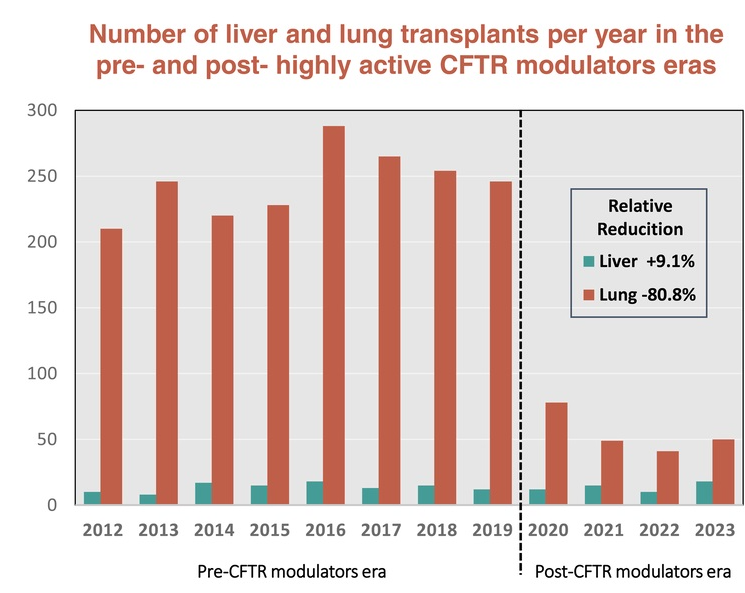
- Impact of CFTR Modulators on the Need for Liver and Lung Transplantation in Patients with Cystic Fibrosis
- Delayed Diagnosis of Cystic Fibrosis and Long-Term Impact
- CHOA Nutrition Support Lecture: Cystic Fibrosis Nutrition -Changing in the Age of ‘Miracle Drug’
- Aspen Webinar 2021 Part 7 -Cystic Fibrosis Liver Disease
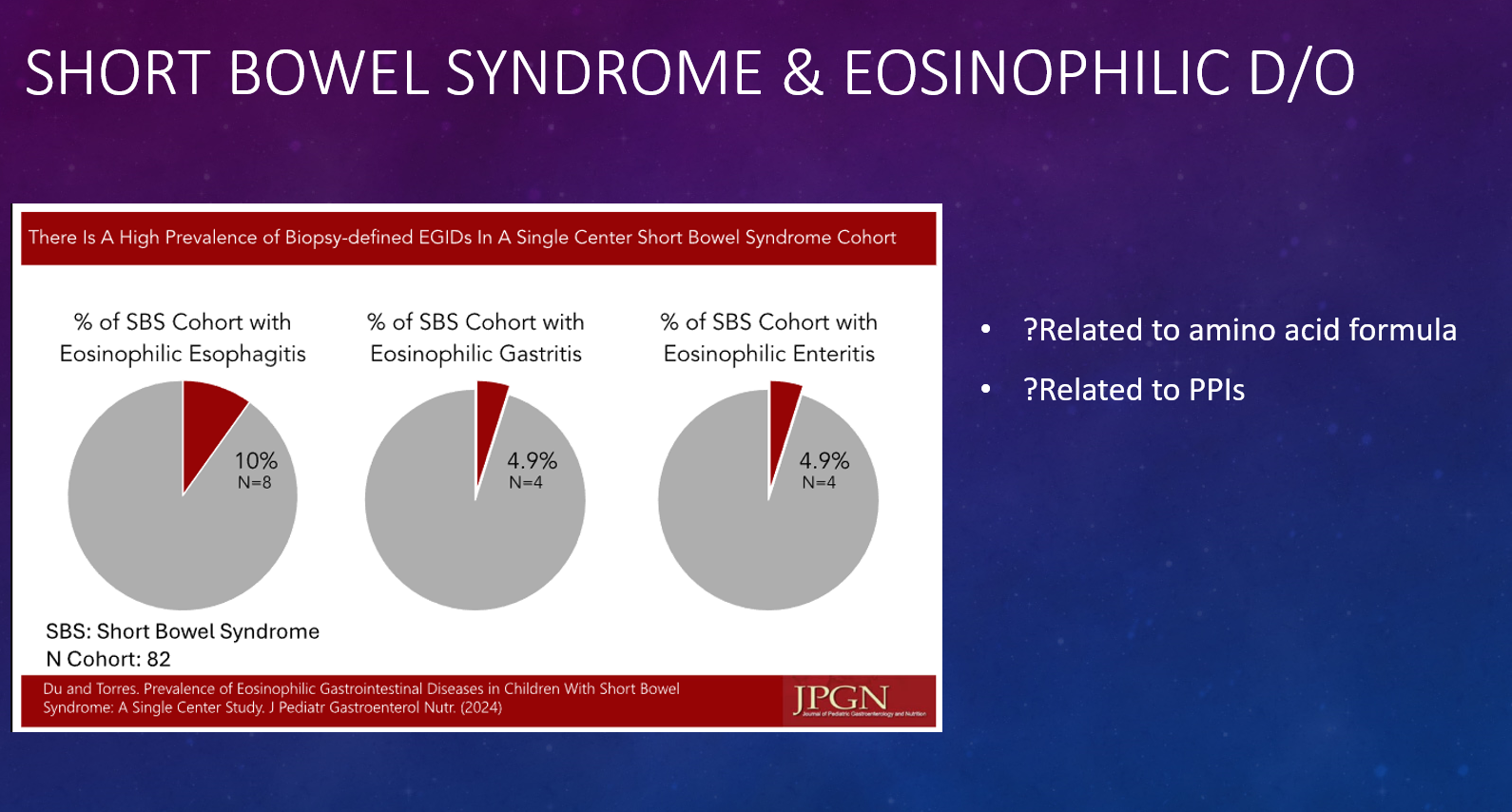
- Intestinal Inflammation in Patients with Cystic Fibrosis
- Data on Immobilized Lipase Cartridge for Patients with CF
- Big Advance for Cystic Fibrosis -Who Will Benefit?