TL Mosher et al. J Pediatr Gastroenterol Nutr. 2026;82:765–769. Guidance adherence in the evaluation and management of hepatic hemangiomas in infants and children
Background: Hepatic hemangiomas are one of the visceral hemangiomas that can be seen and are categorized as infantile or congenital.1, 2 Infantile hepatic hemangiomas (IHH) develop in the first weeks-to-months of life, reach peak size typically by 3 months and often spontaneously involute without medical management within 1–2 years.3 Congenital hepatic hemangiomas (CHH) are present at birth and may rapidly involute, partially involute, or remain static for life.
Guidelines for screening with imaging and laboratory evaluation have been previously published and highlight differences between IHH and CHH.2 (Reference: Iacobas I, Phung TL, Adams DM, et al. Guidance document for hepatic hemangioma (infantile and congenital) evaluation and monitoring. J Pediatr. 2018; 203: 294-300 e2).” Screening with blood counts, fibrinogen, liver function tests, alpha-fetoprotein (AFP), thyroid function tests (TFTs), should be performed at diagnosis for both types of hemangiomas with frequency of repeat testing depending on whether IHH or CHH is diagnosed.2 Echocardiogram at diagnosis should be considered in both if patients have symptoms of cardiac failure. Serial monitoring with liver ultrasounds for at least 1 year or until stable size and vascularity twice in a row is recommended for CHH whereas continued monitoring with ultrasound until complete involution is recommended for IHH.2“
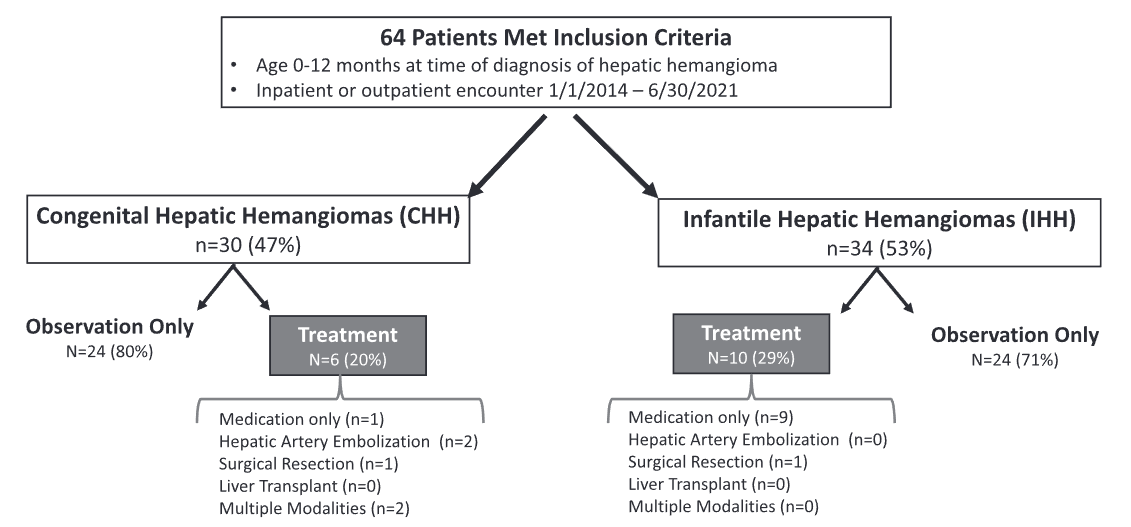
Methods: This study was a retrospective chart review with 64 patients
Key findings:
- 29 (46%) had CHH and 34 (54%) had IHH
- In those 47 patients who were monitored clinically, 62% (n = 29/47) had ALT/AST collected whereas only 51% (n = 24/47) TSH/FT4, 55% (n = 26/47) AFP, and only 26% (n = 12/47) had an INR drawn. Only about one-third (28%, n = 13/47) had a screening echocardiogram performed during evaluation
- When comparing across hemangioma type, patients with CHH were significantly less likely than those with IHH to have TSH/FT4 (26% vs. 75%, p < 0.001) or AFP (39% vs. 71%, p = 0.002) drawn
- Sixteen patients (25%), 6 with CHH and 10 with IHH, required active management.
- Among CHH patients, one (17%) received propranolol prior to transfer to our institution, two (33%) underwent embolization, one (17%) surgical resection, and two (33%) required multiple modalities (i.e., surgery plus embolization). Two patients with CHH receiving multiple treatment modalities developed multi-system organ failure and died.
- Among IHH patients, most patients (90%) received propranolol and one (10%) underwent surgical resection.
Discussion Points:
“Current guidance recommends screening for the complications of hepatic hemangiomas, particularly in patients with diffuse or multifocal hemangiomas, as they are at increased risk for heart failure and hypothyroidism.”
“Current guidance recommends monitoring of hepatic hemangiomas until complete involution of the hemangioma is documented.2, 10 These recommendations are in place given the devastating sequelae that can develop in patients with proliferation of hemangiomas that may be missed when not evaluated with serial abdominal imaging.”
My take: This study shows that about 25% of these patients require medical intervention and is a good reminder of the recommended evaluations.
Related blog posts:
- Natural History and Management of Congenital Hepatic Hemangiomas (this 2025 post has easy algorithm for evaluation)
- Incidental Liver Lesions -What to Do
- Liver Masses -Helpful Reference
- Liver Updates: Statin Protection from HCC, PSVD -new name, novel finding and Hypothyroidism with Hepatic Hemangiomas
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.