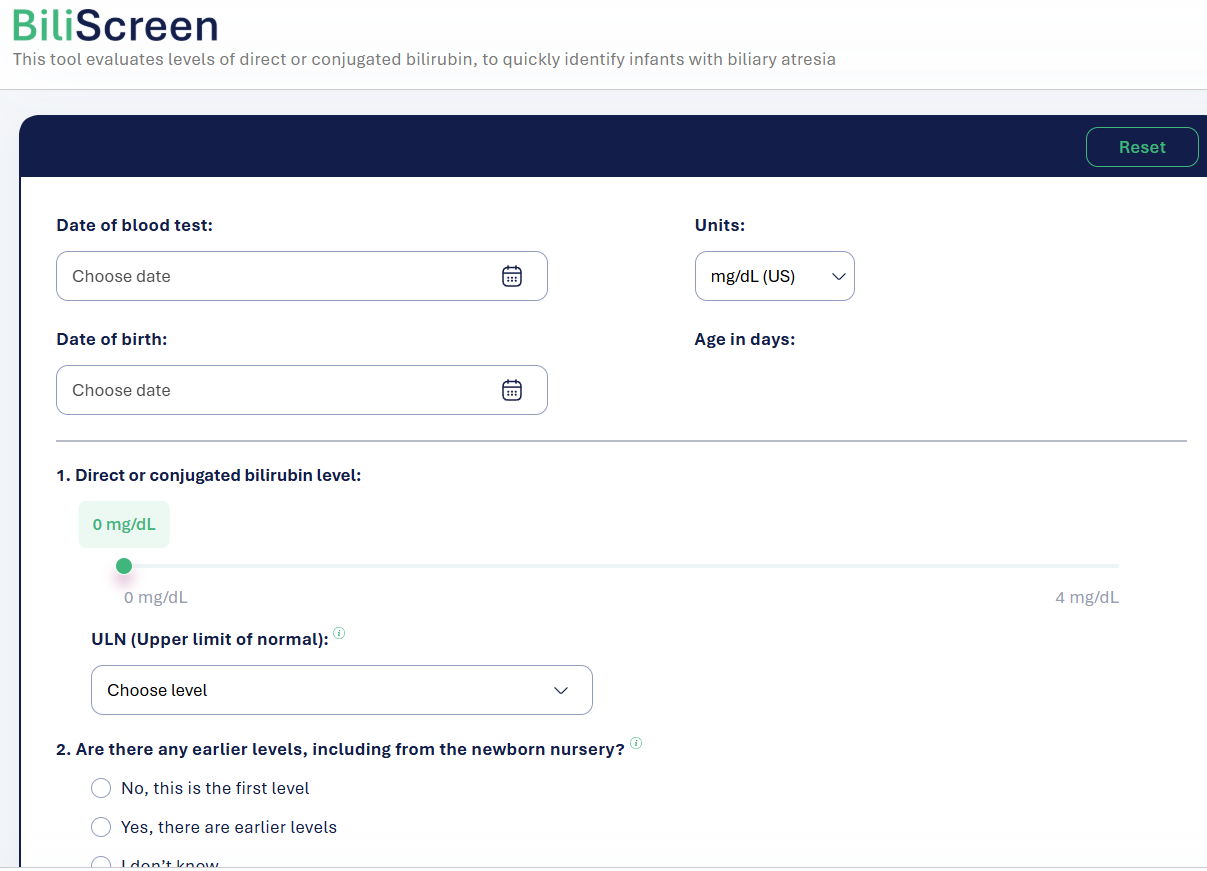
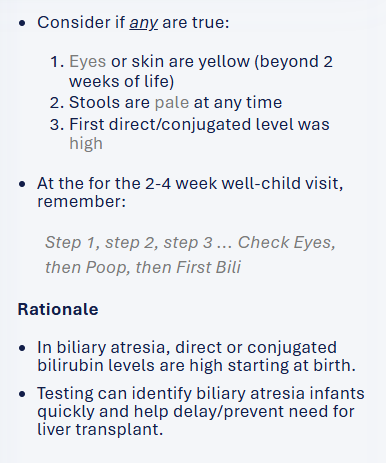
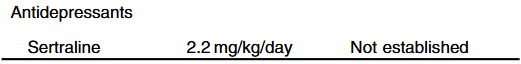There is a new free screening tool, BiliScreen.org, which helps pediatricians triage patients for followup labs and/or hepatology referral. This site incorporates the recent AAP guidelines (see below) for evaluation.
Screenshots from website:
My take:
Overall, this is a quick easy tool which should be helpful & I recommend sharing this website with pediatricians/pediatric health providers
A potential improvement to the site would be a section with an explanation of other causes/consequences of cholestasis which could require more urgent evaluation (eg. metabolic diseases, vitamin K deficiency)
MA Colak et al. J Pediatr Gastroenterol Nutr. 2026;82:358–365. Improvement in bile drainage after Kasai portoenterostomy with a tailored steroid protocol
In this retrospective study, 28 infants underwent Kasai portoenterostomy (KPE) between 2015 and 2025. Group A had 16 infants managed without steroids between 2015 and 2021, while Group B included 12 infants managed under the new tailored steroid protocol between 2021 and 2025.
Determination of bile drainage: Postoperative stool color is monitored closely and collaboratively by hepatologists and surgeons according to the Japanese Tochigi Prefecture 3rd Edition stool card to assess bile drainage over the first five postoperative days.23 Patients with ≥50% of stools at color ≤3 are considered to have poor bile drainage, while those with >50% of stools at color ≥4 are considered to have good bile drainage.
Tailored steroid protocol: “If patients have poor bile drainage, further management depends on age at time of operation. Patients ≤45 days old at operation are started on a combined steroid and antibiotic treatment immediately after bile leak is ruled out using abdominal ultrasound. Patients >45 days old at operation are started on the steroid and antibiotic treatment only if the liver biopsy obtained during operation demonstrated acute inflammation on histology.”
Key findings:
The 3-month post-KPE TB levels were significantly lower in Group B compared to Group A (0.9 [0.3, 1.9] mg/dL vs. 6.5 [0.6, 10.4] mg/dL, p = 0.036)
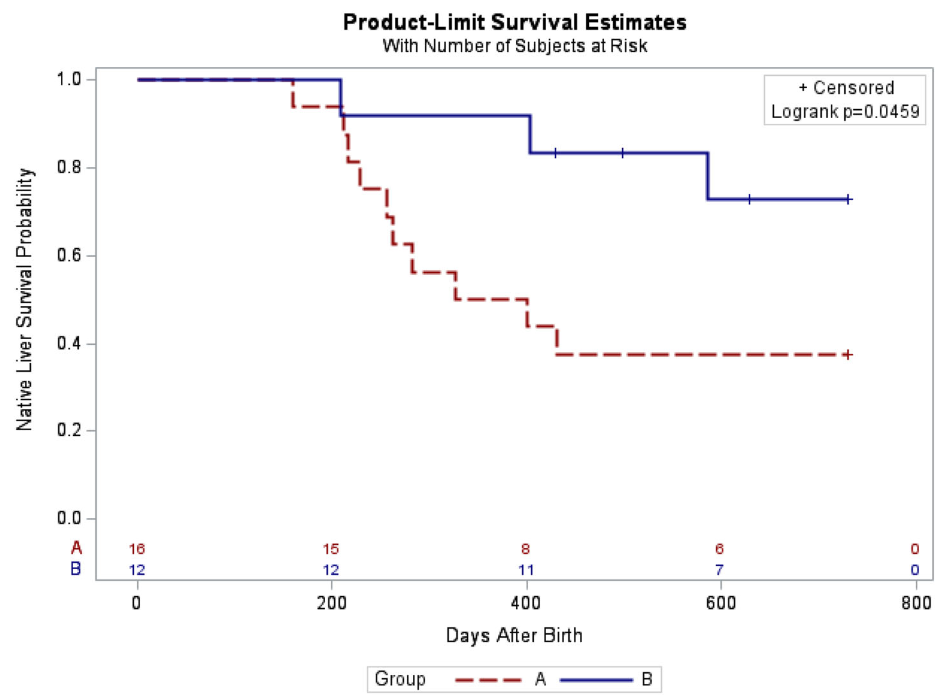
The 2-year native liver survival (NLS) was also significantly higher in Group B (72.9% vs. 37.5%, p = 0.046)
LOS, readmissions, reoperations, and complications in the 90-day postoperative period were not different between both groups
Kaplan–Meier curve of native liver survival at 2 years of age following Kasai portoenterostomy
In their discussion, the authors note that the “multicenter, placebo-controlled, double-blinded steroids in biliary atresia randomized trial (START) included 140 patients from the United States and assessed the effect of high-dose steroids (4 mg/kg/day).16 There was no significant difference in jaundice clearance at 6 months after operation (58.6% vs. 48.6%), nor significant difference in NLS at 2 years of age (58.7% vs. 59.4%) between the steroid and placebo groups.”
Subsequently, “similar to our study, Pandurangi et al. also reported a significant increase in the ratio of patients who had a TB level of <2 mg/dL at 3 months after operation in the customized steroid protocol cohort. However, although the steroid protocol cohort had greater 2-year NLS (68.8% vs. 50%), the difference did not reach statistical significance in their study.”
My take: The START study (n=140), which was powered to detect a 25% absolute treatment difference in TB levels, cannot exclude modest benefits from steroids. This current study, despite its limitations, showed that a tailored protocol for use of steroids may improve outcomes.
START Study: Steroids Not Effective For Biliary Atresia (After Kasai) In this study, the steroid intervention did not affect transplant-free survival which was 58.7% in the steroid group and 59.4% in the placebo group at 24 months of age. In addition, steroids were associated with an earlier onset of first serious adverse events.
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
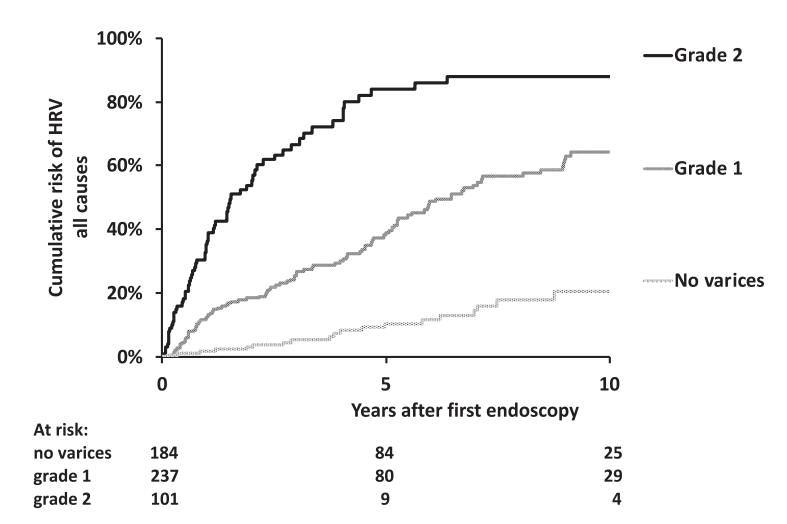
O Ackermann et al. Gastroenterol 2026; 170: 188-198. The Natural History of Gastroesophageal Varices in Children With Portal Hypertension
Methods: Retrospective review of 1586 children with portal hypertension. 590 had two or more upper endoscopies (403 with biliary atresia).
“For the purpose of this study, and based on our previous experience in children,8,11 the endoscopic pattern associated with a high risk of bleeding (ie., HRV) included grade 3 esophageal varices as well as grade 2 esophageal varices with red color signs or gastric varices (cardia), or both.”
The authors developed a HRV [high risk of varices] score as a composite index calculated as follows: 1 point for grade 1 esophageal varices, 2 points for grade 2 varices, 3 points for grade 3 varices, and 1 point each for the presence of red color signs or GOV1 (HRV score range, 0–5). High-risk varices had an HRV score of 3 to 5.
Key findings:
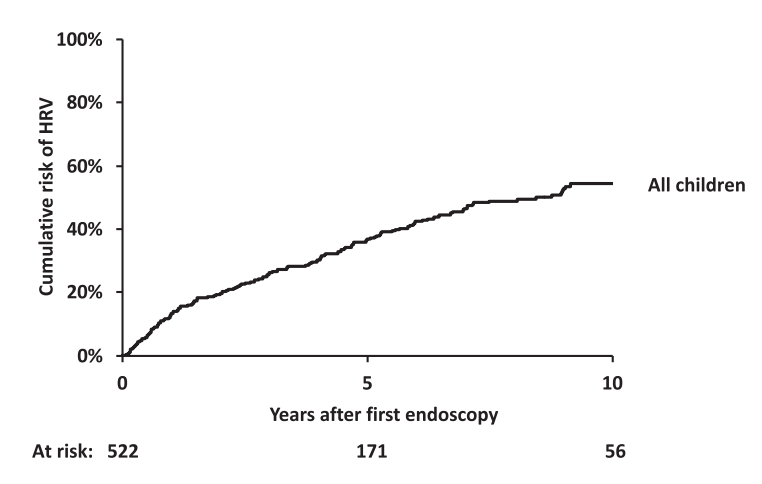
Worsening of the endoscopic pattern occurred in 58% of children over a mean 4-year interval
5- and 10-year probabilities of HRV emergence in initially HRV-negative children were 36% and 54%, respectively
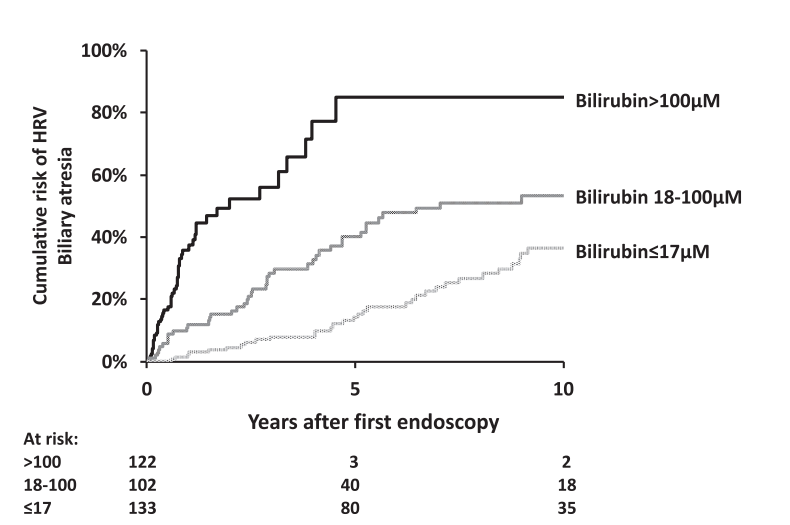
Infants with biliary atresia are at particularly high risk with correlation to the degree of cholestasis (see below)
Platelet count less than 150,000 as an indicator of HRV was mainly useful in older children. “A platelet count of ≥150,000/mm3 was recorded in 205 of the 629 children (32%) with HRV. Moreover, there was a decrease with age in the proportion of children with HRV and a platelet count of ≥150,000/mm3, falling from 62% in children aged <12 months to 2% in patients aged >10 year.” 16% of children 6-8 yrs, 12% of children 8-10 years of age with HRV had platelet count ≥150,000/mm3
“Gastrointestinal bleeding was recorded in 36 of 947 children (3.8%) who did not have HRV at their last endoscopy and in 270 of the 359 children (75%) with HRV at their last endoscopy who did not undergo endoscopic or surgical primary prophylaxis of bleeding.”
Bilirubin of 100 micoMol is equivalent to 5.8 mg/dL and 17.1 is eqivalent to 1 mg/dL
Discussion Points:
Variceal progression was much faster in infants and is is likely due to the severity of cholestasis and its impact on portal hypertension.
“It is notable that children with Alagille syndrome and those with genetic cholestasis with normal GGT have a lower rate of variceal progression and a lower mean HRV score than children with biliary atresia, despite comparably high levels of bilirubin. This suggests that different mechanisms of cholestasis … may have distinct consequences on intrahepatic portal vein branches resulting in varying degrees of portal hypertension.”
“In children with biliary atresia aged <12 months, grade 2 esophageal varices without red color signs or GOV1 (HRV score of 2) should be considered an indication for endoscopic primary prophylaxis.”
“Because the efficacy and safety of β-blockers have not been established in children, we suggest that this pattern—grade 1 varices with red color signs or GOV1—should prompt early repeat endoscopy to detect HRV in a timely manner…this repeat endoscopy could be recommended 6 months after the previous one.”
Limitations: High proportion of children with biliary atresia (limits conclusions with other disorders), and retrospective study since 1990
“Pending the results of future studies, the detection of palpable splenomegaly remains a simple and practical criterion for initiating screening endoscopy in children with portal hypertension”
My take: This is a very useful study providing important data to help improve decision-making in children with portal hypertension.
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
Methods: In this single-center, observational study, the authors deployed an anonymized survey of outcomes that was completed by 107 parents of children with BA who were younger than age 12 years. A detailed assessment of general neurodevelopment (Mullens Scale of Early Learning and Vineland Adaptive Behavior Scale) was carried out in 50 infants younger than 5 years old, and emerging autistic traits (Autism Diagnostic Observation Schedule) were assessed in those eligible. There were 93 matched controls.
Key findings:
Neurodevelopmental concerns were raised by 37% of parents
47% of children required support from at least 1 service (such as speech and language therapy physiotherapy, play therapy, or seen a clinical psychologist), and a further 42% (n = 45) had used more than 1 service. The most accessed service was speech and language therapy (20%, n = 10)
A clinical or research diagnosis of autism was made in 30% of 35 children >2 years old
Early surgical intervention and faster clearance of jaundice after surgery was associated with better general neurodevelopmental outcomes (F = 2.428, P = .042) but not with the presence of emerging autistic traits
My take: High levels of neurodevelopmental difficulties occur in children with BA.
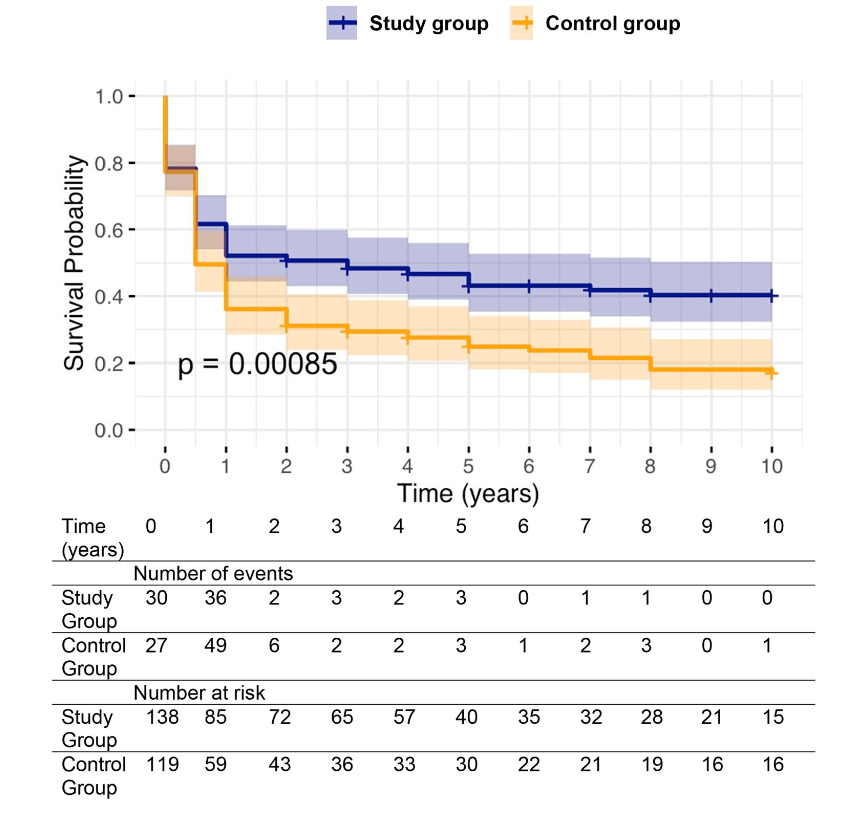
Background: After the START trial in 2014, it seemed that enthusiasm for post-operative steroids for biliary atresia had waned. The START study did not find that steroids improved outcomes after Kasai hepatoportoenterostomy (HPE). Subsequently, though, there have been observational reports of using steroids in a customized fashion to improve outcomes. Langreen et al add to this literature by examining their use of rectal budesonide (2 mg) for 3 months in a retrospective cohort (n=142) with a historical control (n=137). Jaundice-free native liver survival (jfNLS) was assessed at 6 months, 2 years, 5 years, and 10 years post-Kasai.
Key findings:
Improvements were noted in jfNLS at 6 months (53% vs. 39%) , 2 years (45% vs. 22%), 5 years (40% vs. 23%) and 10 years (32% vs. 13%)
These benefits were exclusive to patients with nonsyndromic BA
No serious adverse effects were identified with budesonide
Rationale for rectal budesonide: The authors note that “a single dose of budesonide foam contains about 2 mg of budesonide, equivalent to 25 mg of prednisolone or 20 mg of methylprednisolone…In our series, no serious steroid associated adverse effects were recorded, possibly due to the first pass after rectal administration.”
Limitations: “The retrospective nature of our data analysis allows for variability in the follow‐up protocols, potential biases (historical control group, change of surgeons) and confounding factors cannot be entirely ruled out.”
Kaplan–Meier curve comparing native liver survival between the study and control groups over a 10‐year follow‐up.. Study group—blue. Control group—orange.
My take: The START study with 140 participants was well-designed and did not find a benefit with systemic steroids. However small differences in outcomes can be difficult to identify. Rectal budesonide may improve outcomes. A randomized, double-blind, placebo-controlled trial would be more definitive.
Recently Dr. Balistreri gave our group an excellent lecture. I have taken some notes and shared some slides. There may be inadvertent omissions and mistakes in my notes.
Key Points:
Producing enough bile acids and recycling bile acids in enterohepatic circulation is crucial for bile acid flow. In addition, there are ‘good’ bile acids like cholic acid that have trophic properties and ‘bad’ bile acids like lithocholic acid that cause liver toxicity
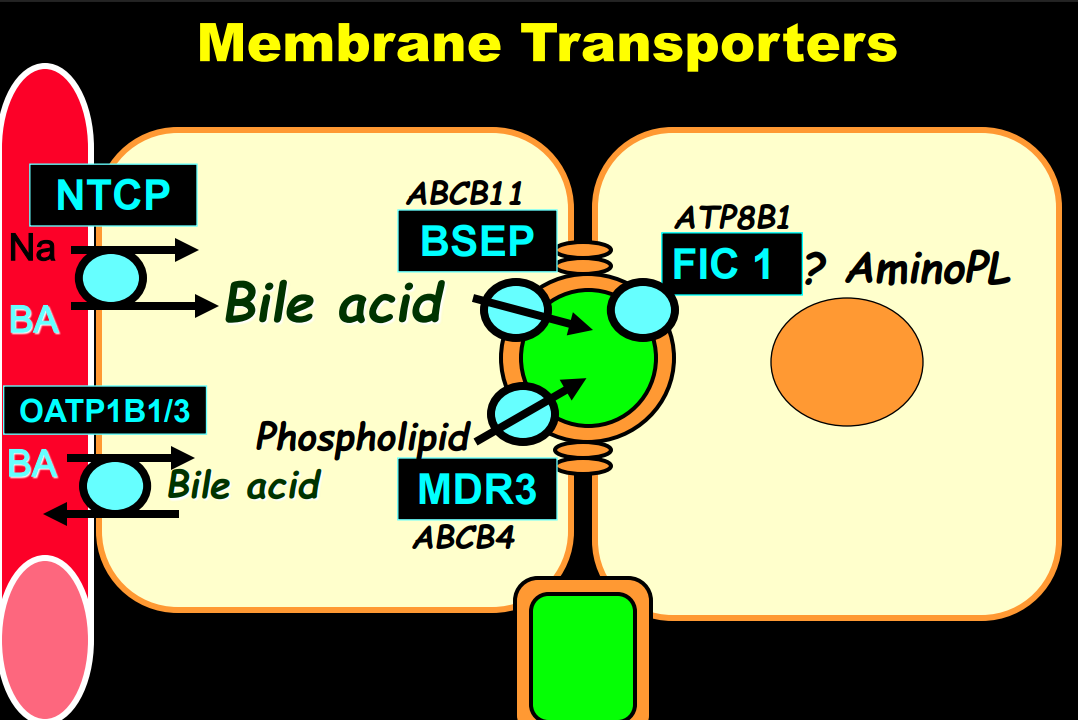
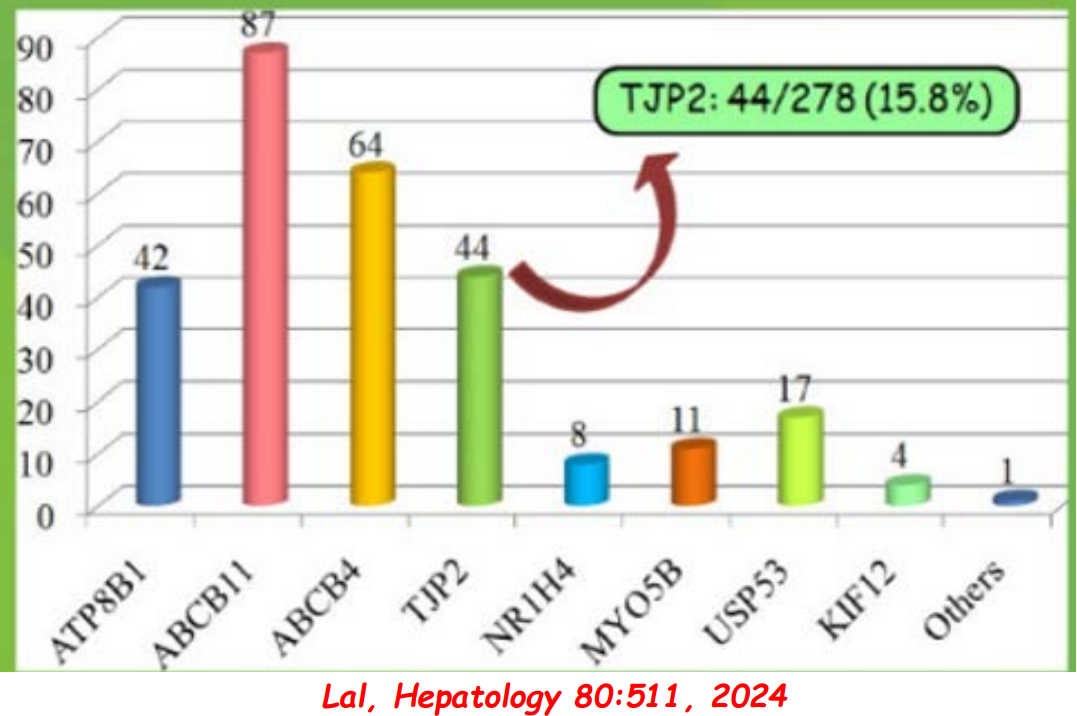
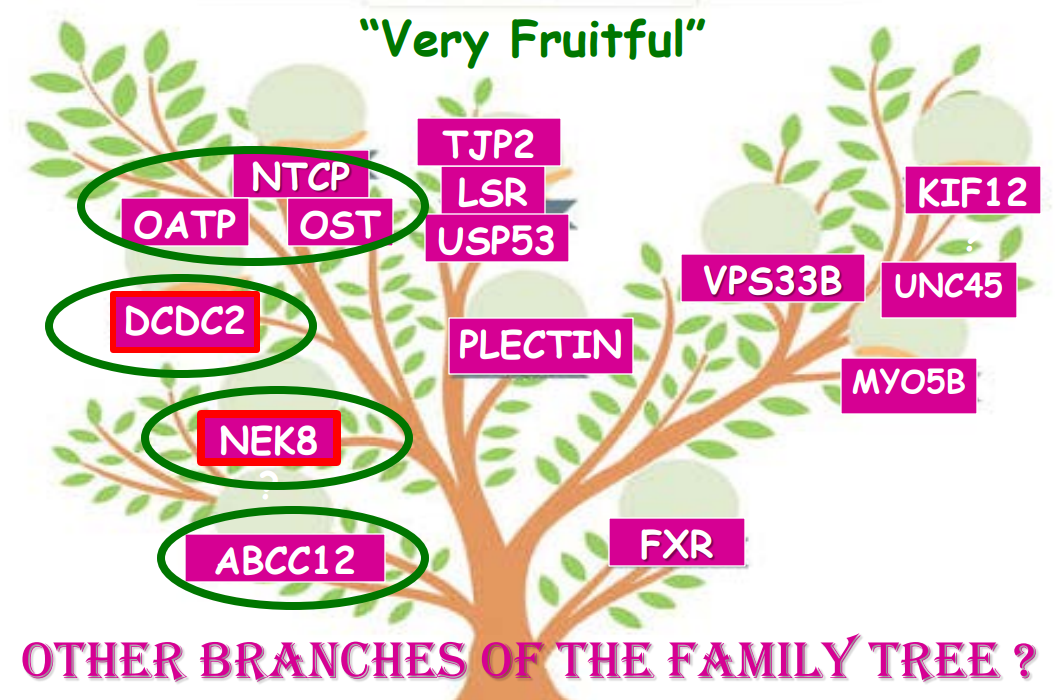
In addition to defects in the metabolic pathway of bile acids, discoveries identified defects in the membrane transporters (eg. FIC1, BSEP, MDR3), trafficking proteins (eg. MYO5B, VPS33B), nuclear control receptors (eg. FXR), and tight junction proteins (eg. TJP2). Tight junction protein defects are associated with bile leakage from bile canaliculus
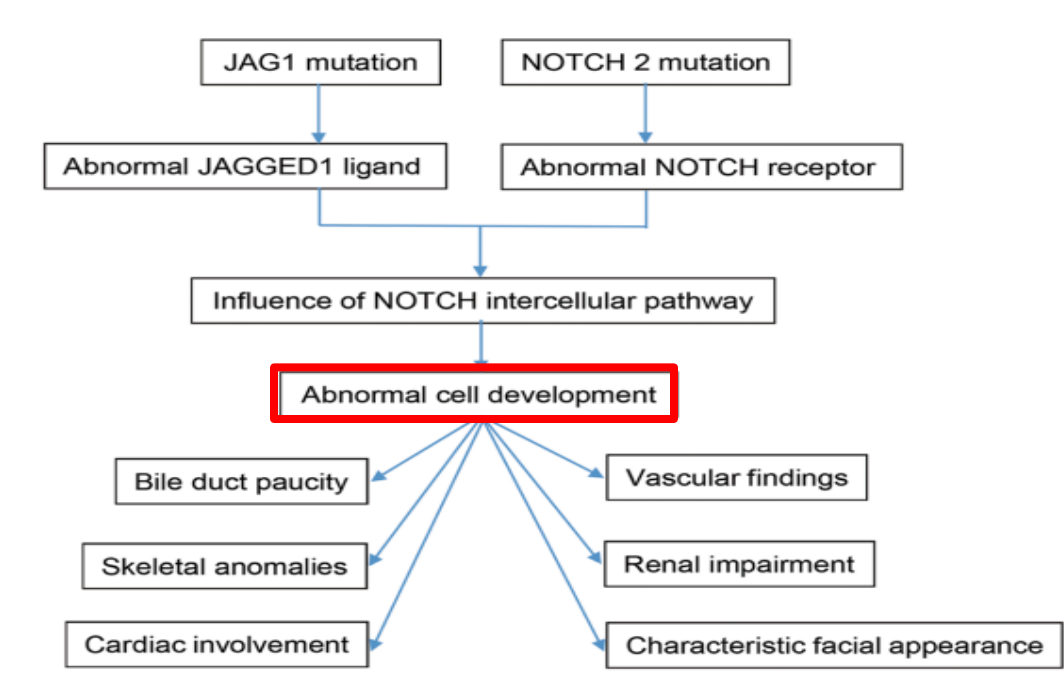
Alagille syndrome, a disorder of embryogenesis, related to JAG1-NOTCH2 signaling pathways affects organs throughout the body
Many of these genetic mutations are now being identified in adults with unexplained liver diseases (eg. intrahepatic cholestasis of pregnancy and cryptogenic cirrhosis)
Cholestasis panels and whole exome sequencing are important tools
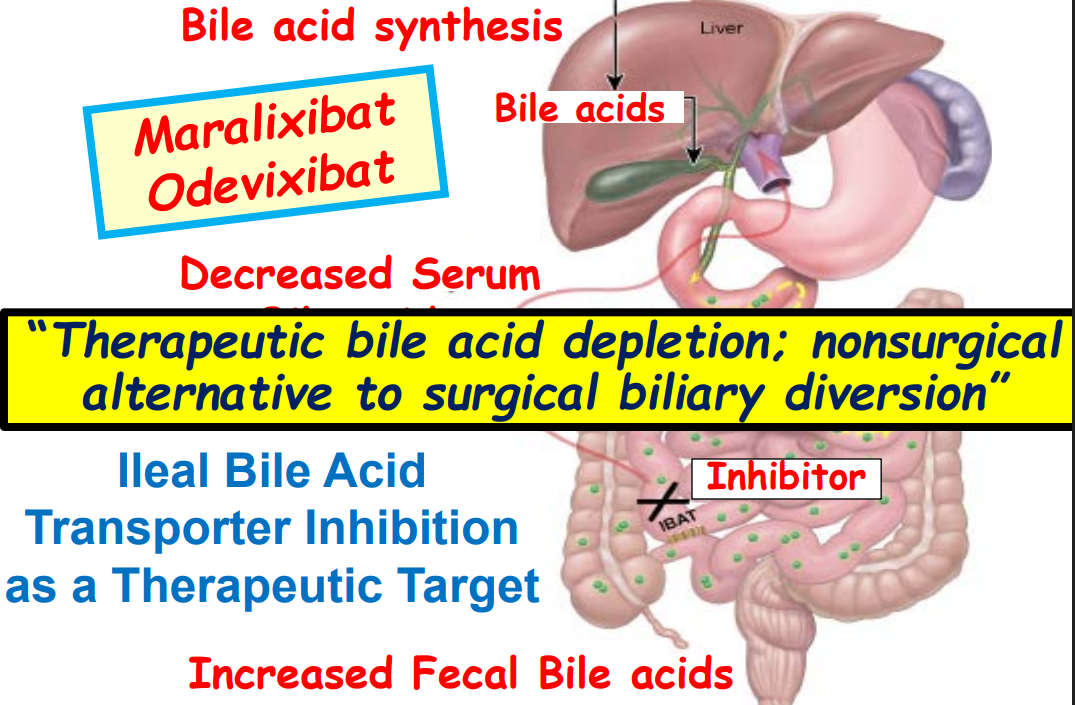
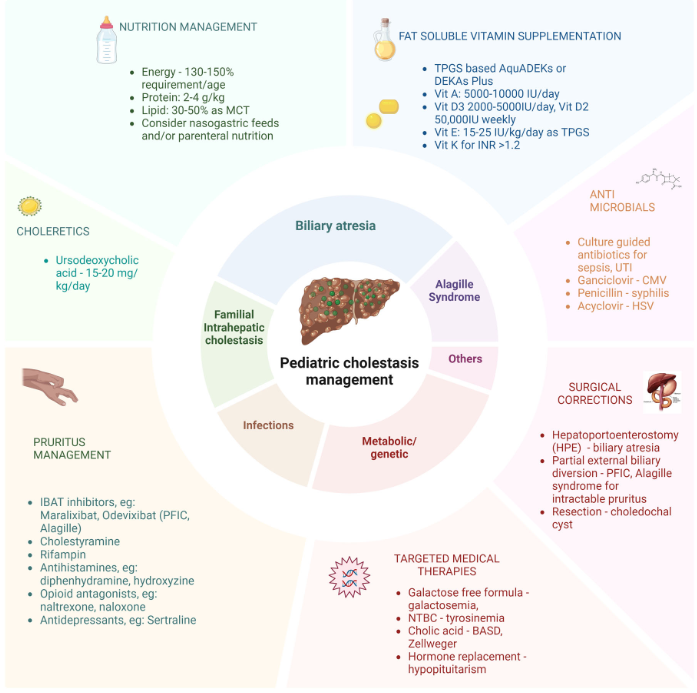
Ileal bile acid transporter (IBAT) inhibitors have emerged as important therapies for conditions like Alagille which were previously treated with biliary diversion
My take: This lecture really shows how the field of pediatric liver disease has been a puzzle. Now one can see how almost all of the pieces of the puzzle work together.
This is a useful review summarizing advances in the management of cholestatic diseases.
Treatment with IBAT inhibitors:
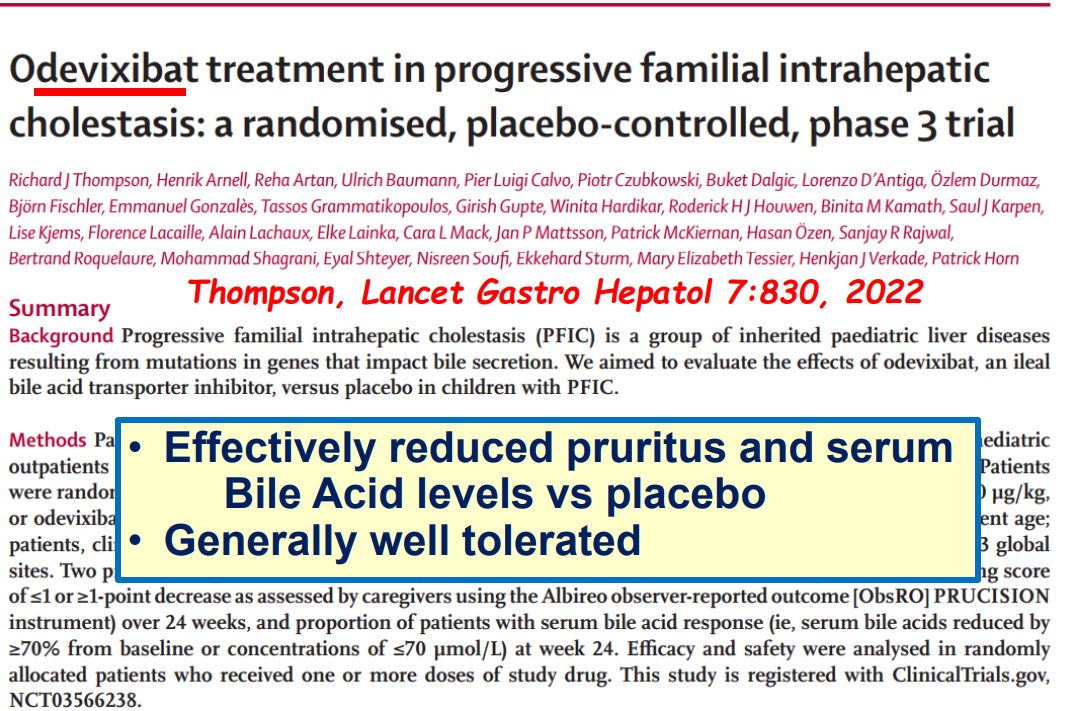
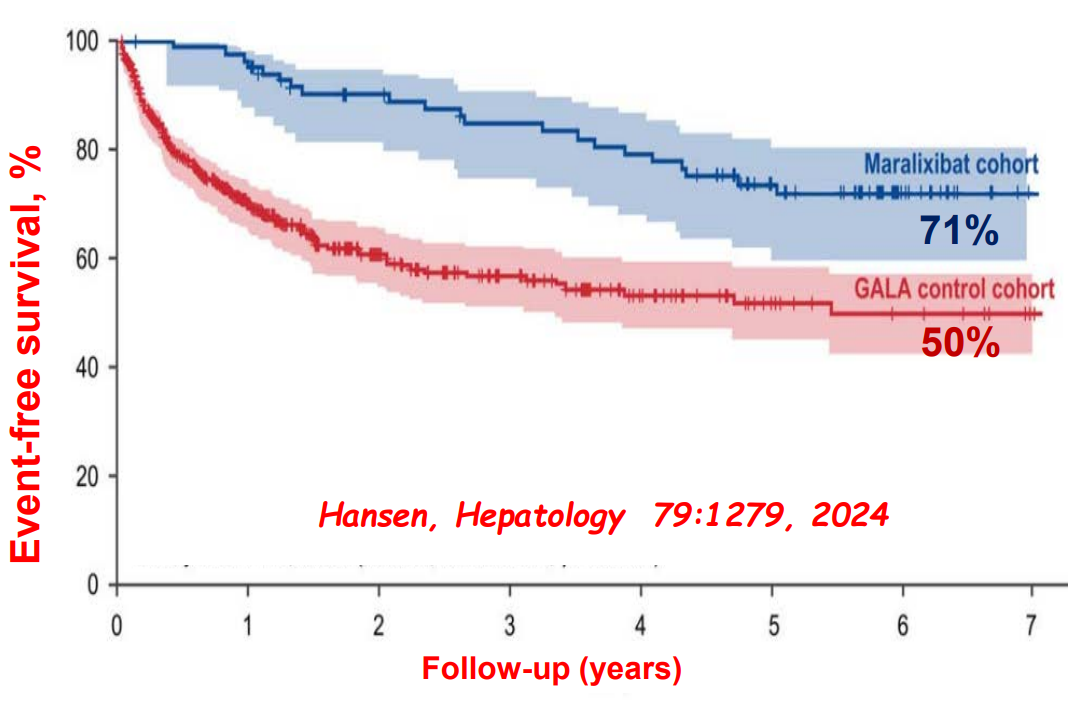
“Improvement in both pruritus and serum BAs/bilirubin levels has been associated with improved event‐free survival and 6‐year transplant‐free survival in ALGS patients treated with maralixibat. Additionally, this class of medication improved overall growth of the patient by improving mean height and weight Z scores that may be related to reduced impact of high serum bile acid levels on the growth axis although further studies are needed to better define the mechanism responsible for this out-come. This finding suggests these parameters could be used as surrogate end‐points for disease severity in diseases like ALGS or PFIC, where the time course to develop the need for LT commonly occurs over many years.”
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
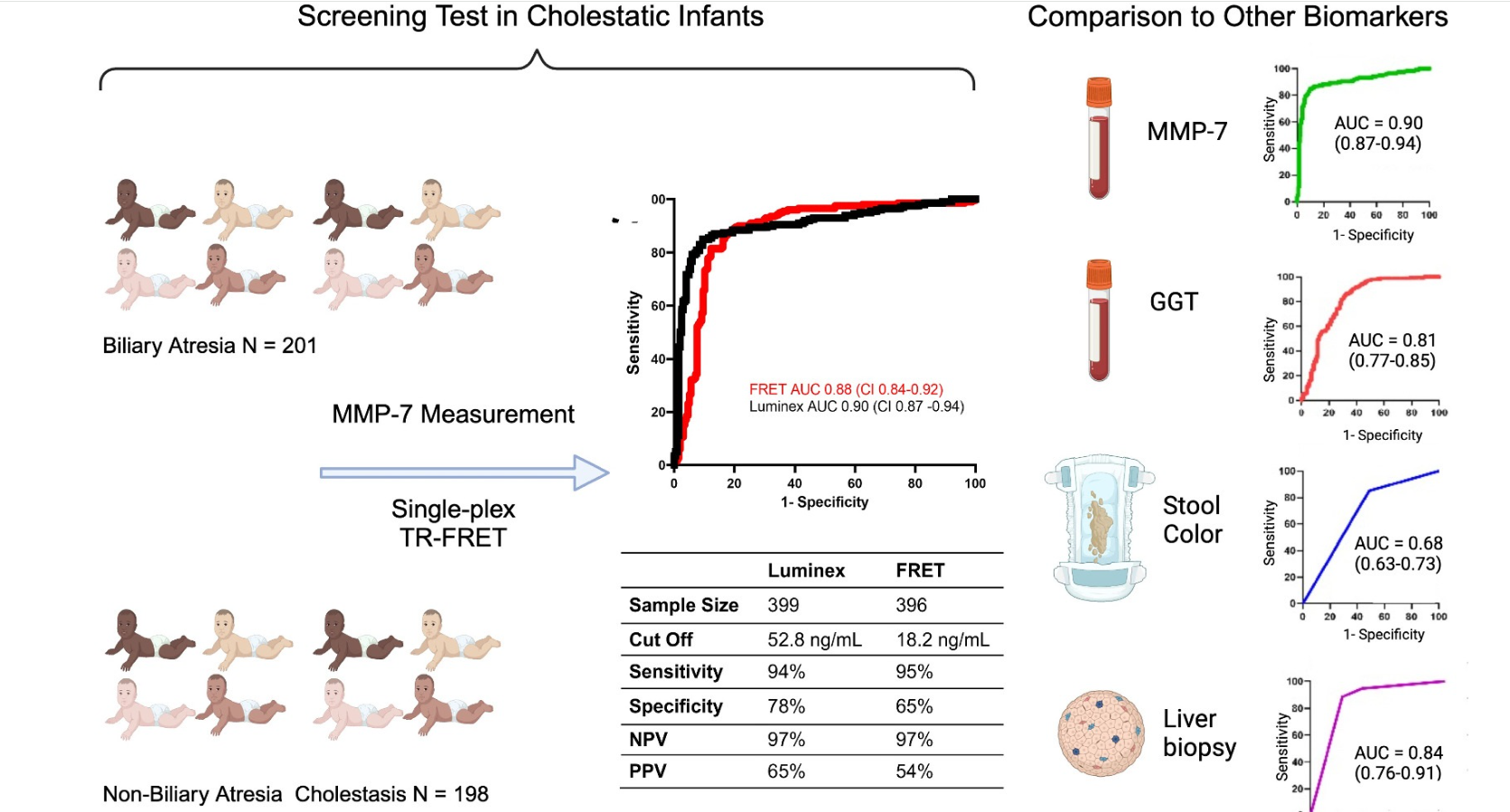
Methods: MMP-7 was measured in serum samples of 399 infants (North America)18 with cholestasis in the Prospective Database of Infants with Cholestasis study of the Childhood Liver Disease Research Network, 201 infants with BA and 198 with non-BA cholestasis (age median: 64 and 59 days, p = 0.94). MMP-7 was assayed on antibody-bead fluorescence (single-plex) and time resolved fluorescence energy transfer assays.
Key findings:
On the single-plex assay, MMP-7 generated an AUROC of 0.90. At cutoff 52.8 ng/mL, it produced sensitivity = 94.03%, specificity = 77.78%, positive predictive value = 64.46%, and negative predictive value = 96.82% for BA.
MMP-7 outperformed other parameters. AUROC for gamma-glutamyl transferase = 0.81 (CI: 0.77–0.86), stool color = 0.68 (CI: 0.63–0.73), and pathology = 0.84 (CI: 0.76–0.91). Obstructive features on pathology were the second-best predictor of BA.
GGT cutoff was 267.5 U/L (per personal communication with senior author) with sensitivity of 86.6%, and specificity of 77.4%
Similar results were found with TR-FRET assay with cut-off of 18.2 ng/mL.
6% (False-negatives) of BA patients had MMP-7 levels below the cutoff
22% (False-positives) of non-BA patients had MMP-7 levels above the cutoff. This included 7 of 8 choledochal cyst patients, 8 of 17 with A1AT, and 13 of 98 with indeterminate cholestasis
In the discussion, the authors note that MMP-7 has performed better in studies with Asian populations, MMP-7 could be useful for dried blood spots in newborns, and could be useful as a measure of successful HPE; continued elevation of MMP-7 has been associated with hepatic fibrosis.
My take: This study shows that MMP-7 is not a perfect assay but often quite helpful. The exact cutoff depends on the specific assay that is utilized. Also, this study shows that checking for A1AT and checking an ultrasound to exclude choledochal cyst need to continue to be done early in the evaluation process.
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
F Koutny et al. JPGN 2024;https://doi.org/10.1002/jpn3.12194. Open Access! Poorly controlled pediatric type 1 diabetes mellitus is a risk factor for metabolic dysfunction associated steatotic liver disease (MASLD): An observational study
Study population, n=32,325. Key finding: Inadequately controlled T1D (HgbA1c >11%) was associated with a higher hazard ratio ((HR: 1.54) of elevated ALT values compared to children with controlled T1D over an observation period extending up to 5.5 years. When both elevated HbA1c (>11%) and overweight were present, the HR was 2.71.
————————————————————————————————————————–
2. G Indolfi et al.JPGN 2024;78:957–972. ESPGHAN recommendations on treatment of chronic hepatitis C virus infection in adolescents and children including those living in resource-limited settings
Summary of Recommendations:
————————————————————————————————————————–
3. F Lacaille et al. JPGN 2024; 78:1374–1382. Awareness, referral and age at Kasai surgery for biliary atresia in Europe: A survey of the Quality-of-Care Task Force of ESPGHAN
Key finding: Data from 785 infants diagnosed with BA from 2015 to 2019 from 18 centers in 15 countries revealed a mean age at referral to tertiary center of 55 days (similar to results obtained in Europe 10–30 years earlier)
This single center retrospective study compared 20 consecutive infants underwent hepatoportoenterostomy (HPE) (beginning in 2017) for biliary atresia (BA) to a historical cohort. Analysis of successful biliary drainage 3 months after HPE (defined as serum total bilirubin (TB) <2 mg/dL) was the primary endpoint; survival with native liver at 2 years was the secondary endpoint.
Protocol:
Cefoxitin was administered to all infants following HPE for 3-4 days.
Standard protocol: If the stool color normalized (pigmented), the infant received “conventional” treatment with trimethoprim-sulfamethoxazole cholangitis prophylaxis, fat-soluble vitamin supplementation with DEKAsPlus or AquaADEKs (1 mL daily), and ursodeoxycholic acid (5 mg/kg twice daily).
Customized protocol: If the stools were acholic (or not consistently pigmented) and </=45 days, the infants received intravenous cefoxitin or piperacillin-tazobactam and methylprednisolone, initial dose 5 mg/kg/day and decreased by 1 mg/kg/day each day thru day 5; then orally treated with dose dropped 0.25 mg/kg weekly. When switched to oral steroids, IV antibiotics were stopped and infant was placed on amoxicillin-clavulanate which was continued until TB <2 mg/dL or discontinuation of corticosteroids (whichever came first).
If stools were acholic and infant was >45 days, then the same treatment was given if there was liver inflammation on histology.
Key findings:
8 had pigmented stools after HPE and received standard protocol.
12 had acholic/inconsistent stools. All of those >45 days had liver inflammation; thus, all 12 received the customized protocol. Two infants had two cycles of steroids/antibiotics who had initial response to treatment and then worsened.
Sixteen of 20 (80%) infants had successful bile drainage, compared to 8 of 20 (40%) infants in the historical cohort (P = 0.0225)
Among the sixteen who have reached two years of age, 11 (68.8%) are alive with native livers versus 10 of 20 (50%) in the historical cohort (P = 0.0970). This did not achieve statistical significance.
The authors established their protocol based on data from Kings College in 2016 suggesting that steroids appeared effective in younger patients who underwent HPE prior to 45 days (Peg Surg Int 2016; 32: 193-200). The START study showed no significant improvement in biliary drainage between patients receiving corticosteroids and placebo. However, in the group <70 days, 72% of infants receiving corticosteroids achieved biliary drainage compared with 57% of the placebo group (P=0.36).
My take: This is a small sample size. Perhaps, this protocol will help improve outcomes. If so, we still don’t know which factor is more important —the IV antibiotics or the high dose steroids. If these agents are helpful, are there other predictive factors –microbiome? MMP-&?
START Study: Steroids Not Effective For Biliary Atresia (After Kasai) In this study, the steroid intervention did not affect transplant-free survival which was 58.7% in the steroid group and 59.4% in the placebo group at 24 months of age. In addition, steroids were associated with an earlier onset of first serious adverse events.