NE Peters et al. NEJM 2024; 391; 56-59. Normalization of C1 Inhibitor in a Patient with Hereditary Angioedema
“An infant with genetically confirmed hereditary angioedema and low C1 inhibitor levels (but without previous episodes of angioedema) underwent liver transplantation for biliary atresia, an unrelated condition. Liver transplantation led to normalization of the C1 inhibitor level and function. To our knowledge, this represents the first patient to be potentially cured of hereditary angioedema.” This case report shows that liver-directed therapy can reverse hereditary angioedema.
Related blog posts:
- Overlooking Important Detail$ in Hereditary Angioedema Treatment
- A “Swell Diagnosis” (part 2)
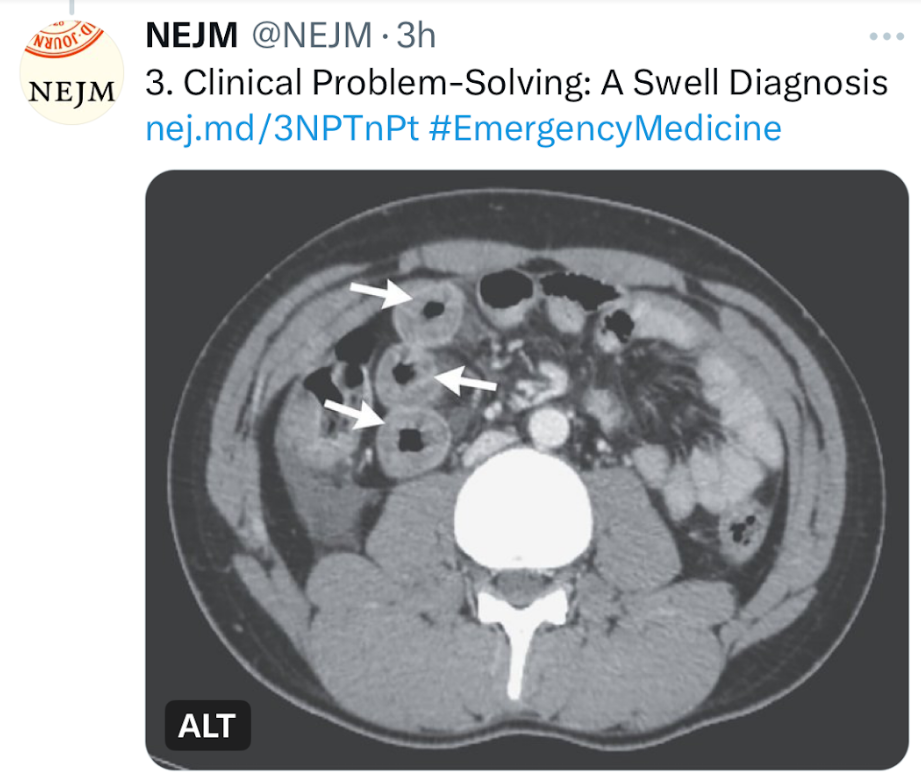
- “A Swell Diagnosis”
- Intermittent Abdominal Pain and Intestinal Swelling –a Mystery?
T Asselah et al. NEJM 2024; 391:133-143. Bulevirtide Combined with Pegylated Interferon for Chronic Hepatitis D
Key finding: At 24 weeks after the end of treatment, HDV RNA was undetectable in 17% of the patients in the peginterferon alfa-2a group, in 32% of those in the 2-mg bulevirtide plus peginterferon alfa-2a group, in 46% of those in the 10-mg bulevirtide plus peginterferon alfa-2a group, and in 12% of those in the 10-mg bulevirtide group.


My take: This is a long (48 weeks) and difficult treatment (2 injection meds and lots of peginterferon side effects). However, there is a fairly good response rate.
Related blog post: Image Only: World Hepatitis Day Infographic
ZM Sellers et al. Hepatology 2024; 79(5):p 1220-1238, May 2024. Open Access! Cystic fibrosis screening, evaluation, and management of hepatobiliary disease consensus recommendations
This link to the article also has links to related AASLD guidelines (eg. management of portal hypertension). Table 2 summarizes the ~34 recommendations which include yearly evaluation with labs, ultrasound at least every 2 years in pediatric patients (age 3 yrs and older) and against routine use of ursodeoxycholic acid.