Recently, Dr. William Balistreri presented a review of some of the biggest advances in pediatric hepatology this past year on the Bowel Sounds Podcast (with the award-winning hosts).
He discussed the following:
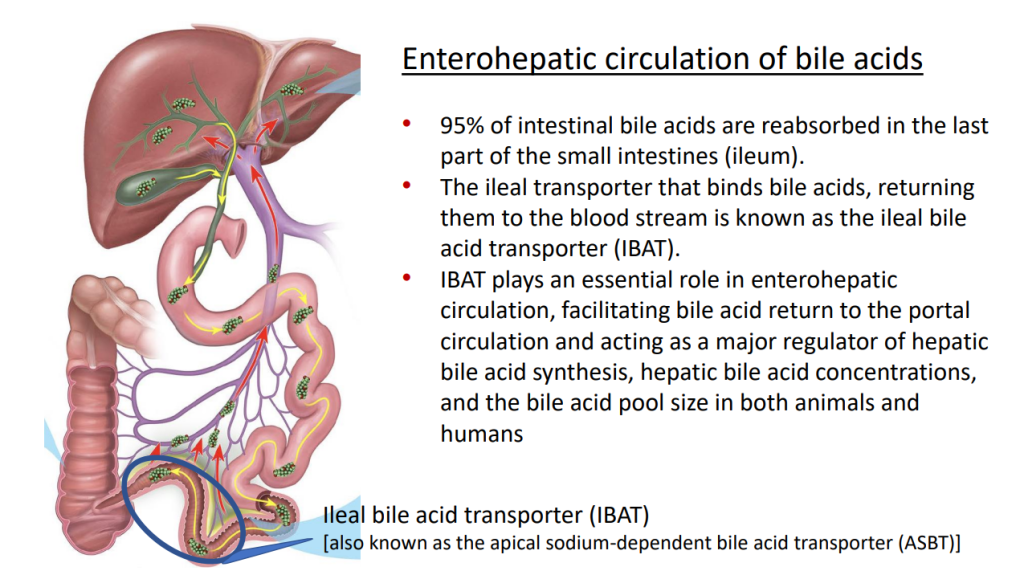
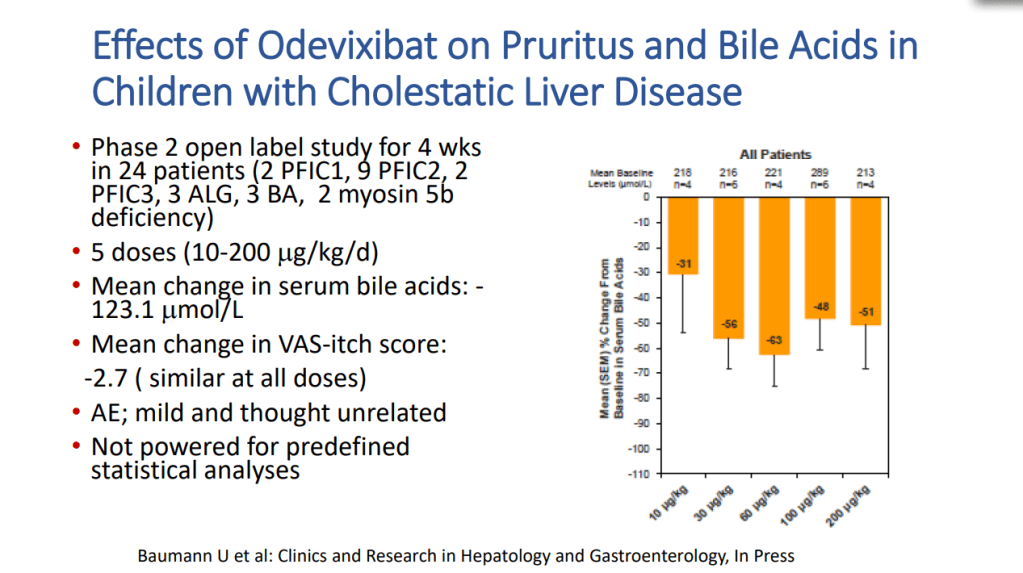
- IBAT inhibitors which are a game-changer for pruritic cholestatic disorders like Alagille syndrome. By reducing itching, it may help many avoid liver transplantation
- HCV medications which usually result in a cure with typical therapy courses running 8-12 weeks
- Emergence of a new treatment, Fazirsiran, for alpha-one antitrypsin deficiency (see blog post below)
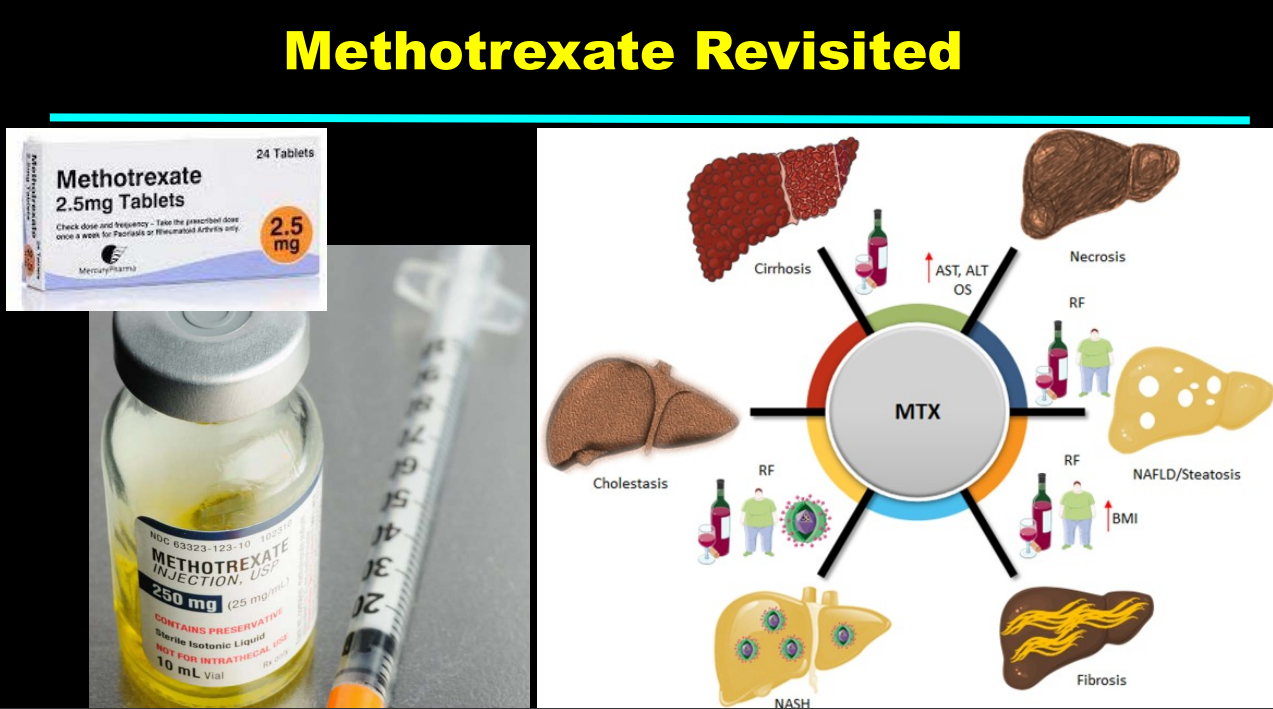
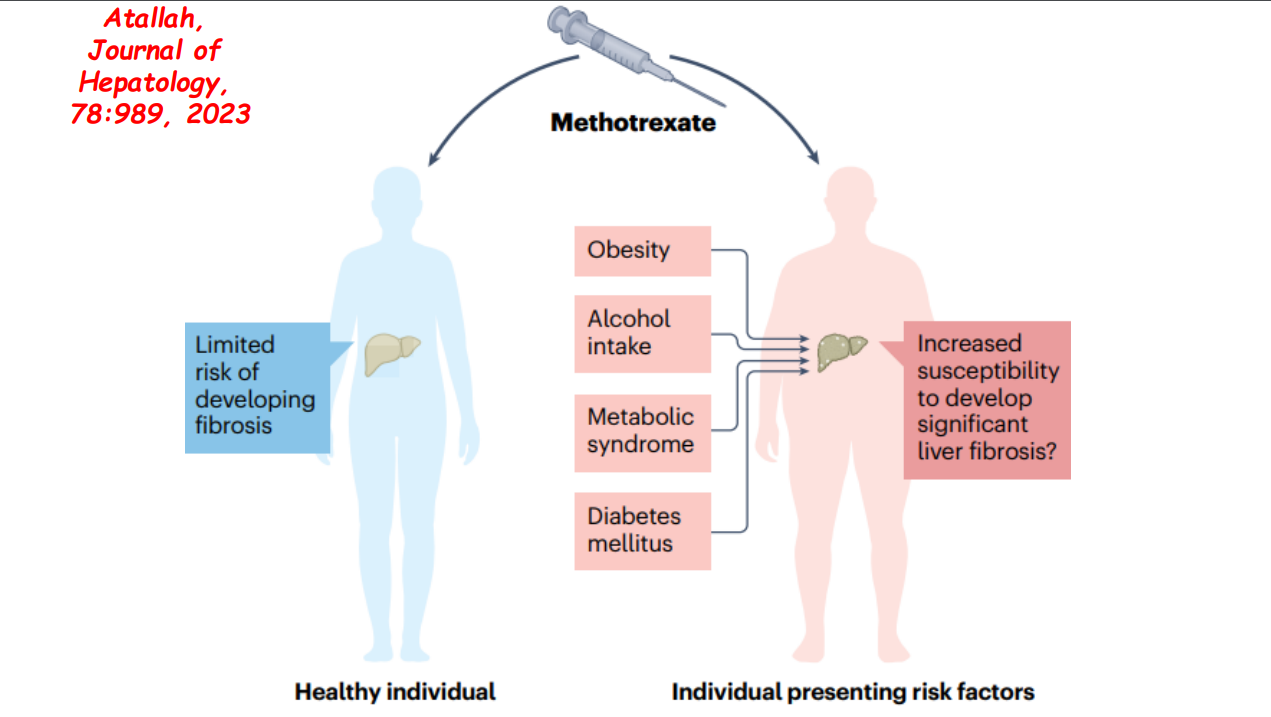
- More data showing the good liver safety of methotrexate in individuals without preexisting liver disease. Dr. Balistreri and colleagues showed pediatric patients with JRA did not develop liver fibrosis/clinical liver disease in 1997.
- How Gilbert’s may be beneficial –>hopeful news for the mildly jaundiced children that we see. Science (M Leslie, 6/8/23): Can ‘toxic’ bilirubin treat a variety of illnesses
He kindly agreed to send me a few slides on the later two subjects at my request:










Related blog posts:
IBAT Inhibitors:
- Six Year Data for IBAT Inhibitor Treatment for Alagille Syndrome
- Lecture: IBAT Inhibitor for Alagille Syndrome
- Aspen Webinar 2021 Part 3-IBAT Inhibitors
- GALA: Alagille Study
Hepatitis C
- Eradicating Hepatitis C Dependent on Congressional Action
- Hepatitis C is Undertreated in the U.S.
- Why CDC is Drafting New Guidelines for Screening Children for Perinatally-Acquired Hepatitis C Infection
- NASPGHAN Foundation: Hepatitis C in Children and Adolescents Lecture Slides
- Improvement in Liver Fibrosis with DAA Treatment of Hepatitis C in Adolescents
- Hepatitis C in 2020: NASPGHAN Position Paper
- Online Aspen Webinar (Part 4) -How to Treat Hepatitis C in Children
- Medical Progress: Toward Hepatitis C Elimination Detailed information on treatment of HCV
- Oral Pan-Genotypic HCV Drugs Approved For Children Starting at Age 3 Years
Alpha-One Antitrypsin
- RNA Interference (Fazirsiran) for Liver Disease Associated with Alpha-1-Antitrypsin Deficiency
- Alpha-1-Antitrypsin Deficiency (review May 2020). 35% of adults with ZZ genotype show clinically-significant liver fibrosis. Risk factors for advanced fibrosis: male gender, metabolic syndrome/obesity, and alcohol consumption.
- Liver Shorts March 2020 (A1AT Heterozygosity worsens NAFLD/contributes to cirrhosis)
Dr. Balistreri
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 1)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 2)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 3)
- “The Paramount Health Challenge for Humans in the 21st Century”