More from the following: JR Thiagarajah et al. Gastroenterology 2018; 154: 2045-59. (Senior authors/corresponding authors: Yaron Avitzur and Martin Martin). This article provides an excellent review of persistent infantile diarrhea and provides algorithms to help in the evaluation of these disorders. These algorithms incorporate the role of exome sequencing.
The authors divide infants with watery diarrhea/CODEs into five categories -detailed in their Table 2 which also has OMIM #, inheritance pattern, gene name, protein function:
#1 Epithelial nutrient/electrolyte transport:
- congenital chloride
- congenital sodium
- glucose-galactose malabsorption (GGM)
- primary bile acid diarrhea
- acrodermatitis enteropathica
#2 Epithelial enzymes and metabolism
- Congenital lactase deficiency
- Sucrase-isomaltase deficiency
- Trehalase deficiency
- Enterokinase deficiency
- DGAT1 deficiency
- PLVAP deficiency
- Abetalipoproteinemia
- Hypobetalipoproteinemia
- Chylomicron retention disease
- Dyskeratosis congenita
- Kabuki syndrome
#3 Epithelial trafficking and polarity
- Microvillus inclusion disease
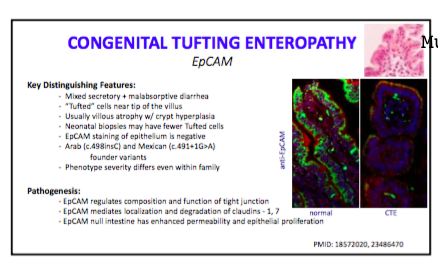
- Tufting enteropathy
- Syndromic Na diarrhea
- Trichohepatoenteric syndrome 1 & 2
- Familial hemophagocytic lymphohistiocytosis 5
- TTC7A deficiency
#4 Enteroendocrine cell dysfunction
- Enteric anendocrinosis
- X-linked lissencephaly and MR
- Proproteint convertase 1/3 deficiency
- Mitchell-Riley syndrome
#5 Immune dysregulation-associated enteropathy (partial list)
- IPEX
- ICOS deficiency
- ADAM17 deficiency
- EGFR deficiency
- CD55 deficiency
- CTLA4 deficiency
- LRBA deficiency
- XIAP
So, to tackle this long list the authors recommend combining typical clinical evaluation along with early genetic evaluation.
Clinical evaluation of watery diarrhea:
- Early endoscopic biopsy (EGD/Flex sig) -obtain samples for routine histology and for electron microscopy. Disaccharidase evaluation can be helpful; though, “these enzymatic assays are often unreliable due to poor sampling or in the setting of inflammation or villus atrophy due to secondary disaccharidase deficiency.”
- If normal villus/crypt architecture, the next step is determining whether the diarrhea improves with fasting. This could indicate GGM, sucrase-isomaltase, congenital lactase deficiency or enteroendocrine cell loss. The first three can be elucidated by offering specific dietary challenges using either a feeding trial with carbohydrate-free or fructose-based formula.
- If normal villus/crypt architecture, and if the diarrhea does not improve with dietary manipulation, consider congenital chloride diarrhea, congenital sodium diarrhea, primary bile acid mediated diarrhea, and hormone-induced diarrhea.
- If normal villus/crypt architecture, and there is hypoalbuminemia/PLE, consider DGAT1 deficiency, CD55 deficiency, and lymphangiectasia.
- If abnormal villus/crypt architecture, then this is likely either a postinfectious/autoimmune disorder or due to an epithelial structural defect like tufting enteropathy, microvillus inclusion disease, TTC7A deficiency or SKIV2L defect
When one looks at the magnitude of disorders that could result in CODEs and their potential clinical importance, it is not surprising that the authors state emphatically:
“In cases of a suspected CODE, where the diagnosis based on clinical evaluation is unclear, it is now standard of care to perform whole-exome sequencing to identify a possible causative genetic mutation.”
My take: This article provides a great deal of information in tackling a difficult problem.
Related blog posts:
Disclaimer: These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.

Little Talbot State Park (near Amelia Island)