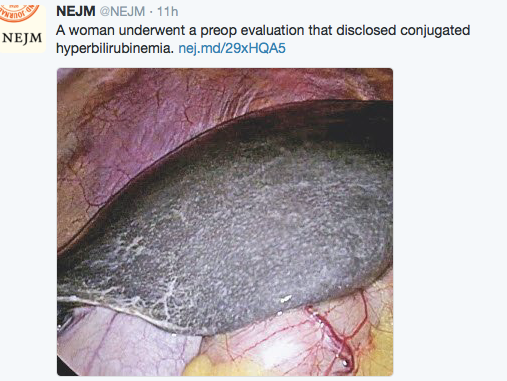
Typically, most pediatric gastroenterologists want to remember that the gross appearance of the liver in patients with Dubin-Johnson is black as this is rumored to be a frequently asked question for board testing. Practically, though other features are important to recognize since the color of the liver is not readily evident except during surgery. As such, a recent retrospective study (T Togawa et al. J Pediatr 2018; 196: 161-7) describes 10 neonatal patients from Japan.
Key findings:
- Only 3 of the 8 patients who underwent liver biopsy had a grossly black liver
- All liver specimens showed no expression of multidrug resistance-associated protein 2 and increased expression of the bile salt export pump protein
- Homozygous or compound heterozygous pathogenic variants of ABCC2/MRP2 (ATP-binding cassette subfamily C member 2/multidrug resistance-associated protein 2) were identified in all patients
The clinical course was similar in the patients:
- Cholestasis self-limited/benign: “severe cholestasis developed in the neonatal period…reaching a maximum at 19 to 60 days. Cholestasis then decreased and disappeared at 2 to 9 months of age.” Acholic stools were common during the cholestatic phase.
- Serum AST and ALT remained consistently normal
- There was no hepatosplenomegaly and no failure to thrive
My take: Dubin-Johnson syndrome is much easier to identify with the availability of genetic panels.
Related blog post: Dubin-Johnson Syndrome