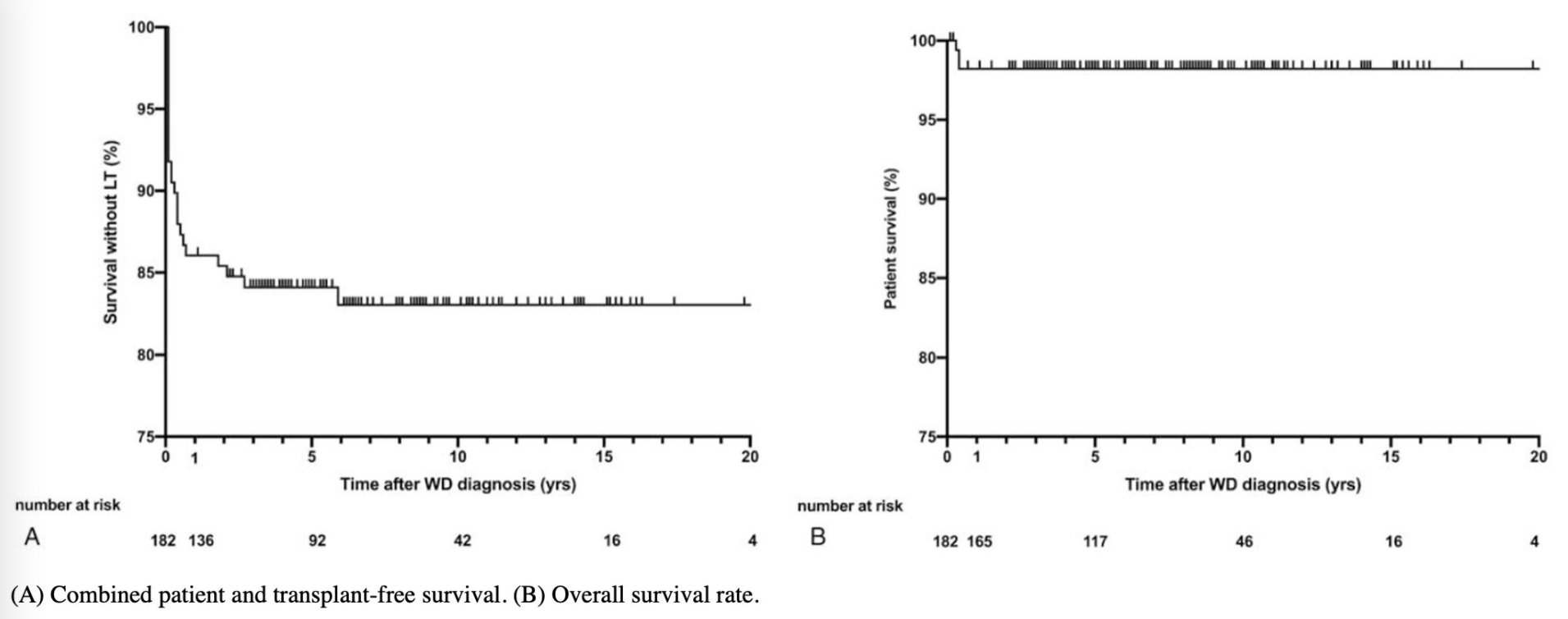
ML Schilsky et al. Hepatology 2023; 77: 1428-1455. Open Access! A multidisciplinary approach to the diagnosis and management of Wilson disease: Executive summary of the 2022 Practice Guidance on Wilson disease from the American Association for the Study of Liver Diseases
This article is an excellent review of Wilson disease (WD). It reviews clinical manifestations, disorders with overlapping findings and recommendations for diagnosis/management. “This executive summary provides a condensed overview, including the clinical algorithms, tables, and full complement of guidance statements.” Though this ‘summary’ is a lengthy publication, “the full Guidance document with comprehensive text, complete references, and supplementary materials (“A Multidisciplinary Approach to the Diagnosis and Management of Wilson Disease: 2022 Practice Guidance on Wilson Disease from the American Association for the Study of Liver Diseases”) is available on the American Association for the Study of Liver Diseases (AASLD) website (https://doi.org/10.1002/hep.32801).”
Some of the recommendations:
The Leipzig score for diagnosis of Wilson disease may aid in diagnosis

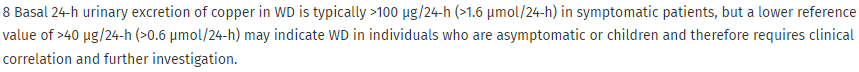



My take: This is a useful guidance and the tables/algorithms should help with both diagnosis and treatment adjustment. In my limited experience with WD, I have had a lot of difficulty with adherence to treatment in the small sample of patients under my care.
Related blog posts:
- Wilson’s Disease –Pediatric Guideline
- How Effective is Zinc Therapy for Wilson’s Disease?
- Liver Shorts -February 2021 (part 1)
- New Way to Diagnosis of Wilson’s Disease: ATP7B Peptides | gutsandgrowth
- Finding the Right Specialist | gutsandgrowth This post has link to AASLD guidelines for Wilson disease.
- Data on Chelators for Wilson Disease | gutsandgrowth
- “This Is A Stick Up — Your Money or Your Life”