Yesterday, I listed the posts with the most views. The posts below were the ones I like the most.
General/General Health:
- William Meyers
- The Health Consequences of Climate Change
- David Brooks: “Kindness is a Skill”
- No Exaggeration: Too Many Children are Dying in the U.S.
- High Toll of Sudden Infant Death
Nutrition:
- The Paramount Health Challenge for Humans in the 21st Century
- Food Safety Lecture –It’s Still a Jungle Out There
- Ensuring Safe Infant Formula Use –More Complicated Than You Think
Liver:
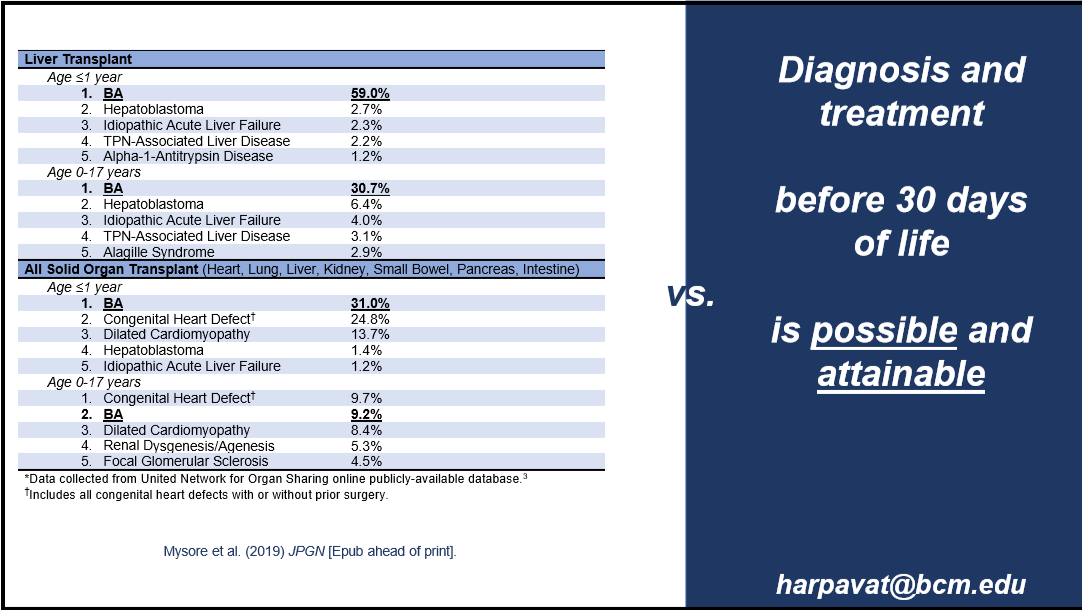
- Blood Test is Better Than a Liver Biopsy for Biliary Atresia
- #NASPGHAN19 Liver Symposium (part 3)
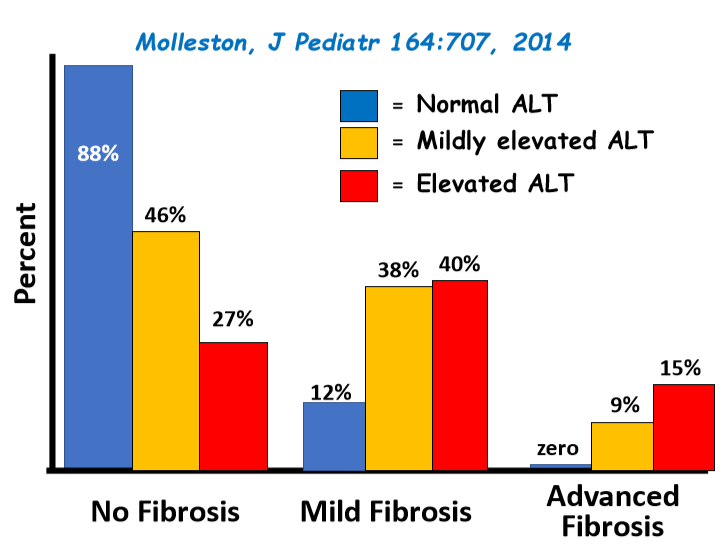
- Low Free Sugar Diet for NALFD
Endoscopy:
- Our Study: Provider Level Variability in Colonoscopy Diagnostic Yield
- #NASPGHAN19 Postgraduate Course (part 3)
Intestinal Disorders:
- Safety of Senna-Based Laxatives
- Management of Acute Severe Colitis
- Dietary Therapy for IBD
- Oral Antibiotics for Refractory IBD (link on page for post with full text of article: October 2019 IBD Shorts)
- PPIs: Good Safety News
- Aprepitant for Cyclic Vomiting
- ESPGHAN Recommendations for Juvenile Polyposis Syndrome (position paper)
- How (Un) Helpful is AD Manometry in Children with Orthostatic Intolerance?

Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.