A Phillip et al. J Pediatr Gastroenterol Nutr. 2025;81:913–921. A narrative review of the ileal pouch in pediatric inflammatory bowel disease and familial adenomatous polyposis
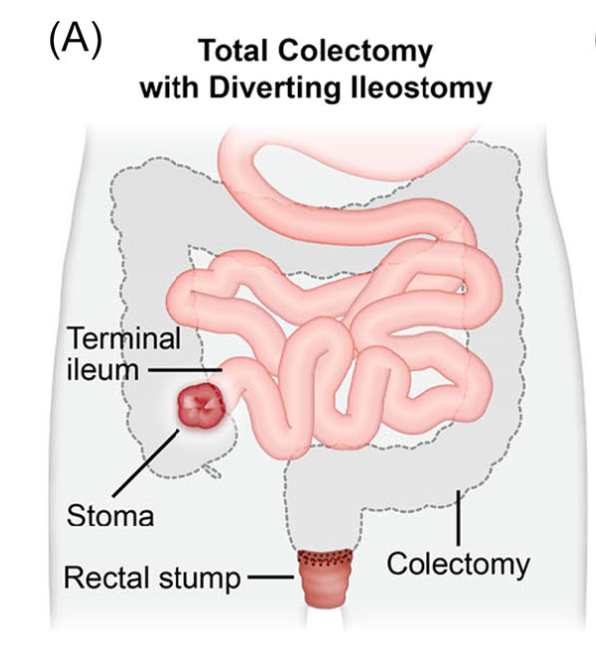
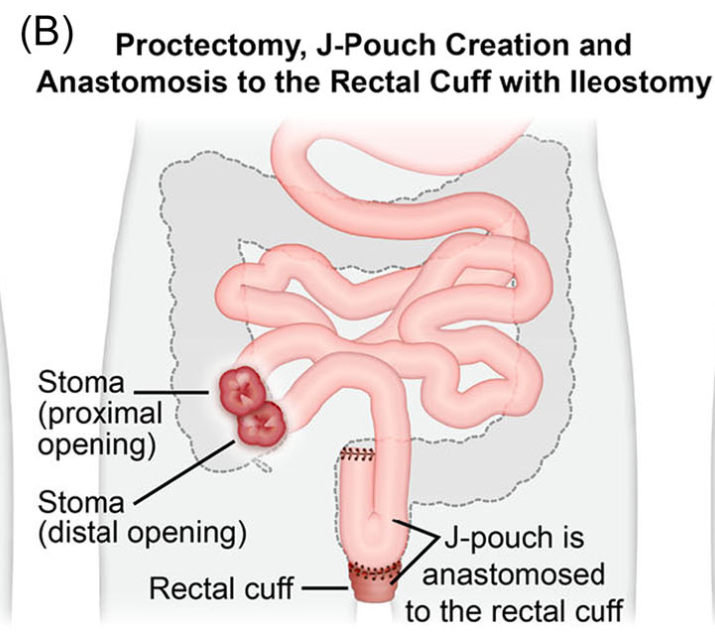
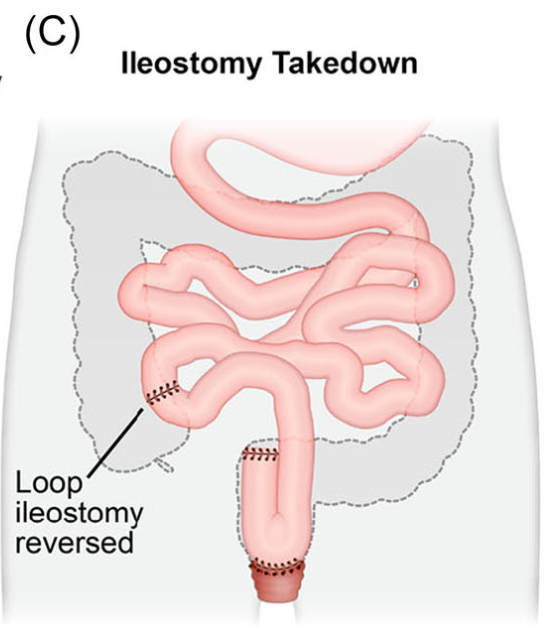
Introduction: Total proctocolectomy with ileal pouch-anal anastomosis (IPAA) can be a life changing solution for a subset of pediatric inflammatory bowel disease (IBD) and familial adenomatous polyposis (FAP) patients. For patients with severe disease a three-stage approach is commonly performed.
Creation of IPAA -Three Stages:
Endoscopic Images and IPAA Anatomy:
The article provides guidance on complications including pouchitis, CD-like inflammation of the pouch, J-pouch failure, fertility after IPAA along with follow-up/screening recommendations.
As for screening, adult guidelines recommend annual screening for IBD patients with high risk features—previous dysplasia, primary sclerosing cholangitis, type C mucosa, refractory pouchitis. In those without these features, guidelines are variable, with one suggesting screening every 5 years. In FAP patients, the recommendation for surveillance screening following IPAA is pouchoscopy every 1–2 years.8
My take: Most pediatric gastroenterologists are not proficient in pouch management due to the small number of our patients needing IPAA. This review provides a terrific review/resource.
Recently, Dr. Steve Erdman gave our group a great update on polyposis disorders. My notes below may contain errors in transcription and in omission. Along with my notes, I have included many of his slides.
Key points:
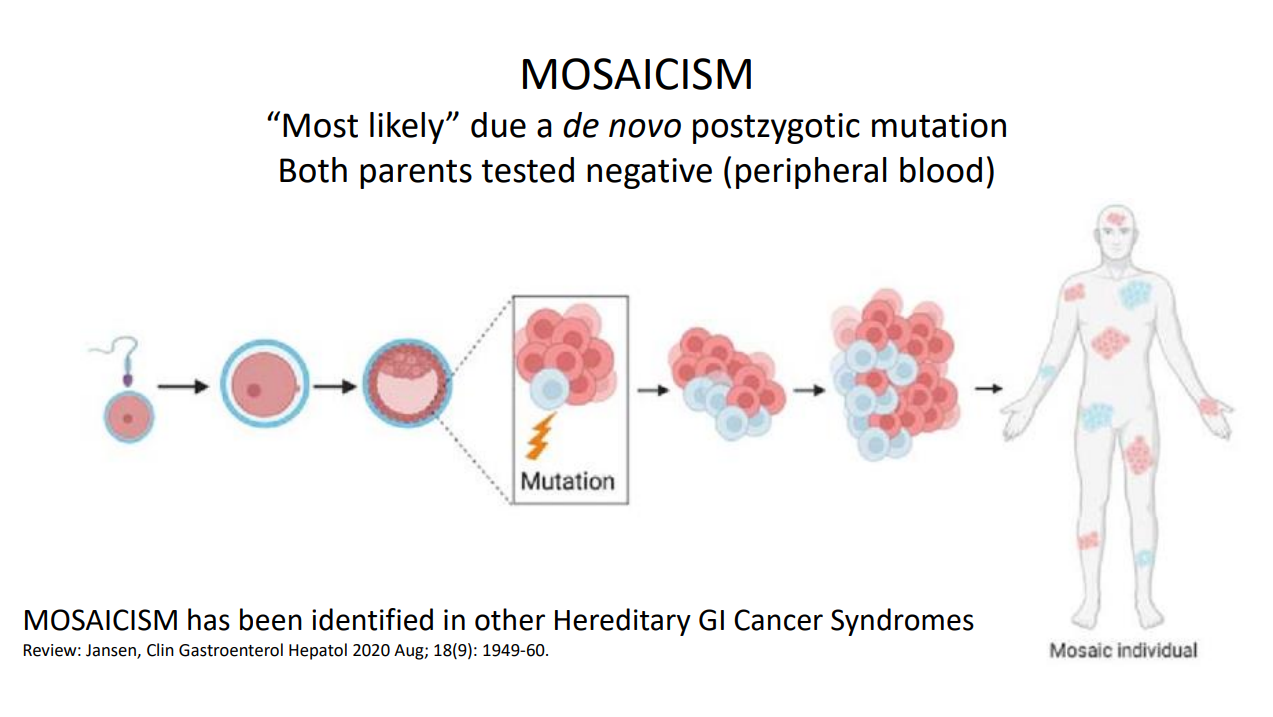
There has been breath-taking progress in understanding of polyposis disorders. It is important to have genetic counselors participate to optimize testing and evaluation
In patients with suspected polyposis syndromes, a genetic diagnosis is very important and can help guide management
Family history is very important. If several family members have had GI or other cancers at a young age, more aggressive interventions are usually indicated. However, individual family members can have a wide variation in presentation
In patients with many polyps, it is worthwhile to alert family to the fact that some polyps can be missed on colonoscopy and to contact medical team if there are recurrent symptoms like rectal bleeding
Some disorders, like juvenile polyposis syndrome (JPS), the connection between polyp presence and cancer risk is not clear. The increased risk for colon cancer my remain after polypectomy.
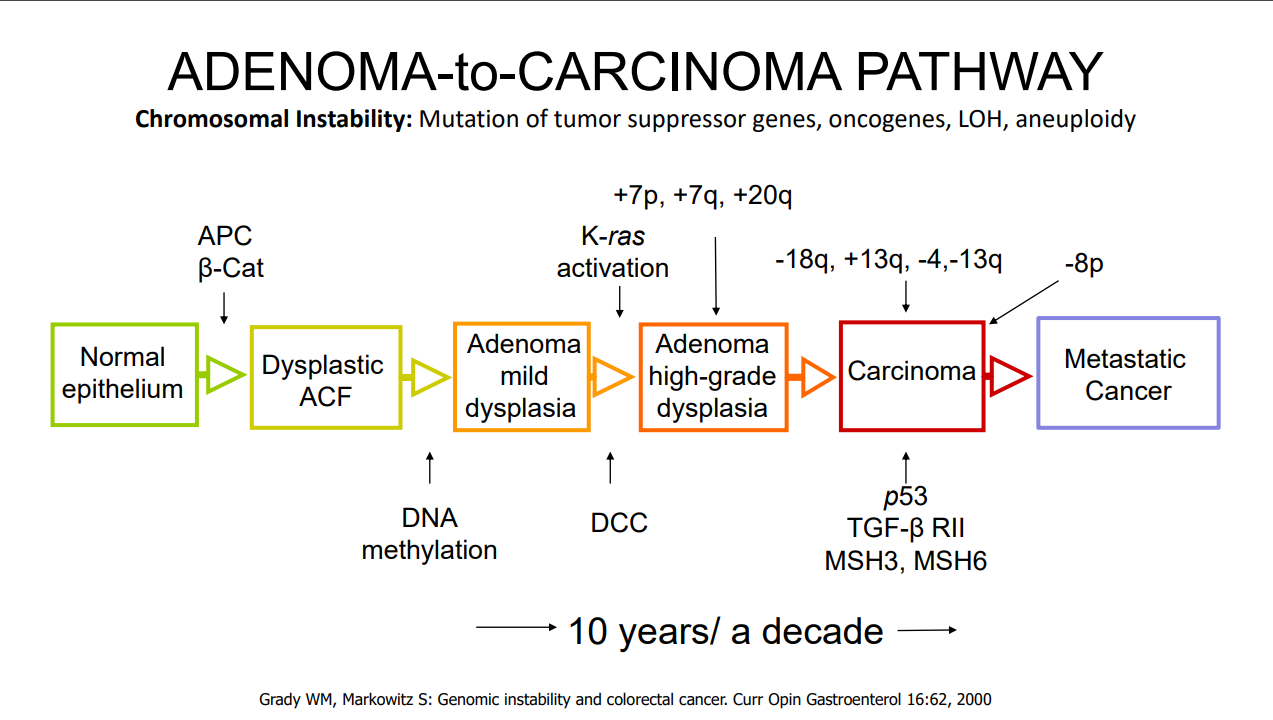
For most individuals with adenomatous polyps, removal of the polyps prevents cancer development (in the GI tract) as there is a well-described adenoma-to-cancer sequence that typically takes 7-10 years to progress from adenoma to colon cancer
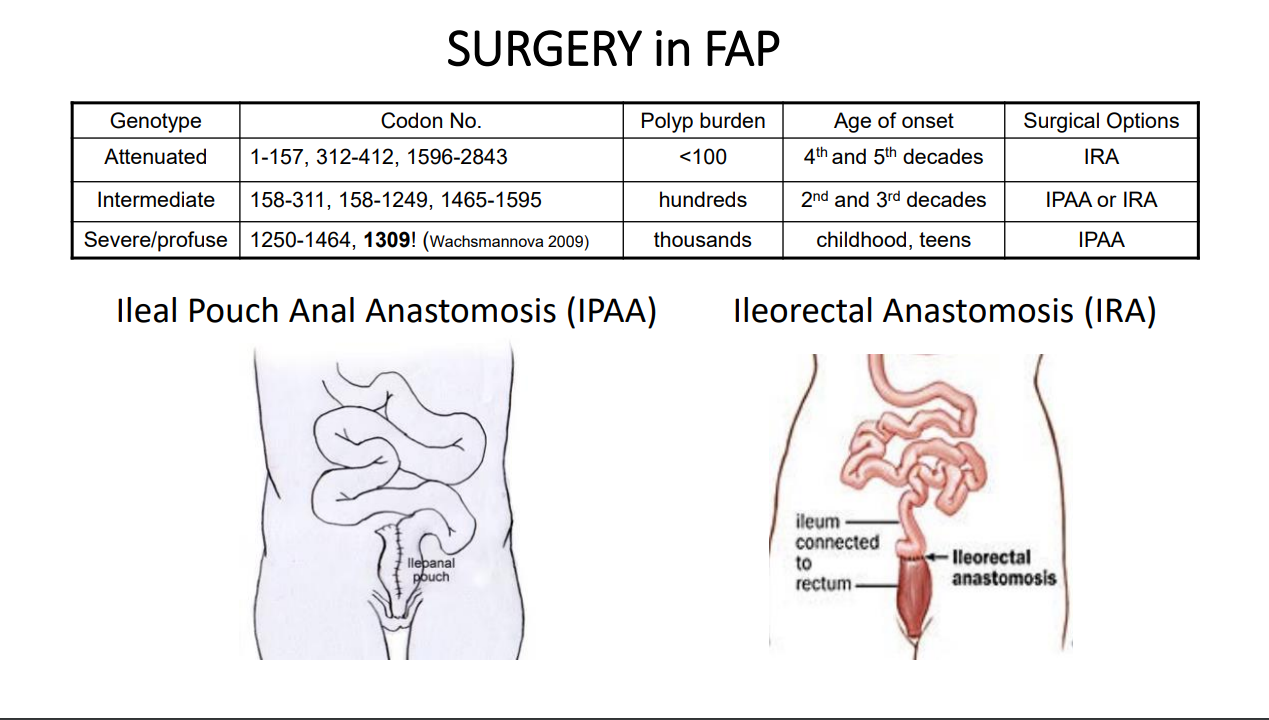
With FAP, the severity is related in part to the specific mutation. Mutations in the mutation cluster region are associated with an aggressive phenotype and mutations causing attenuated FAP are less aggressive
For FAP, timing of potential colectomy involves factors including severity as well as social factors. In teenagers in which there is a concern about being lost to follow-up, this is a factor that could influence earlier intervention
Many times a 2nd opinion in pathology can be helpful, especially if colon cancer is reported. However, histologic dysplasia can be tricky as well
Isolated CHRPE usually does not require evaluation. Dr. Erdman noted that sometimes genetic testing is offered to a family for reassurance. He discouraged colonoscopy in this setting unless a genetic diagnosis has been established or symptoms like rectal bleeding are present. The penetrance of APC mutations (development of polyps) can be quite variable (especially with attenuated form)
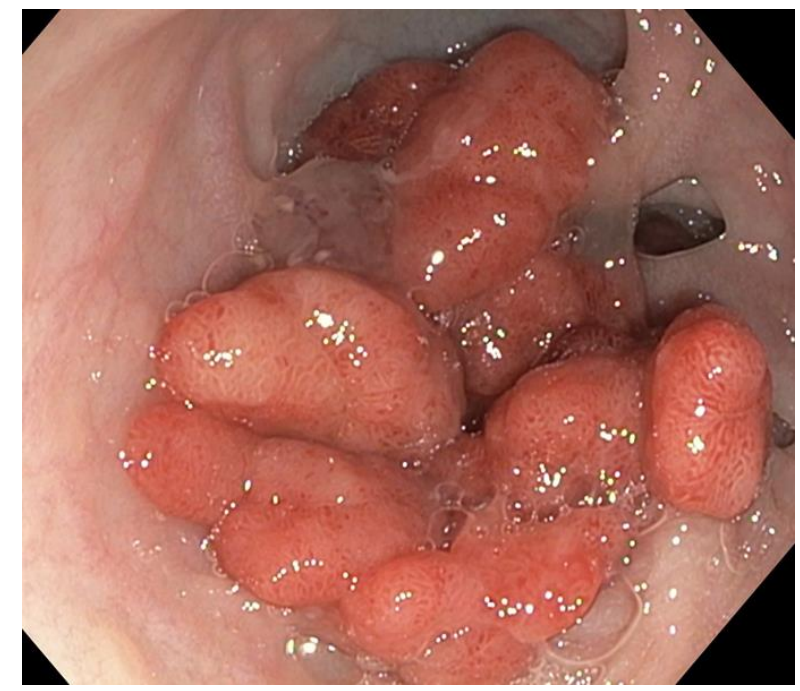
Case #1 presented a 14 yo with 50+ multilobulated pedunculated polyps which histologically were tubulovillus adenomas. Initial diagnosis was elusive despite extensive testing
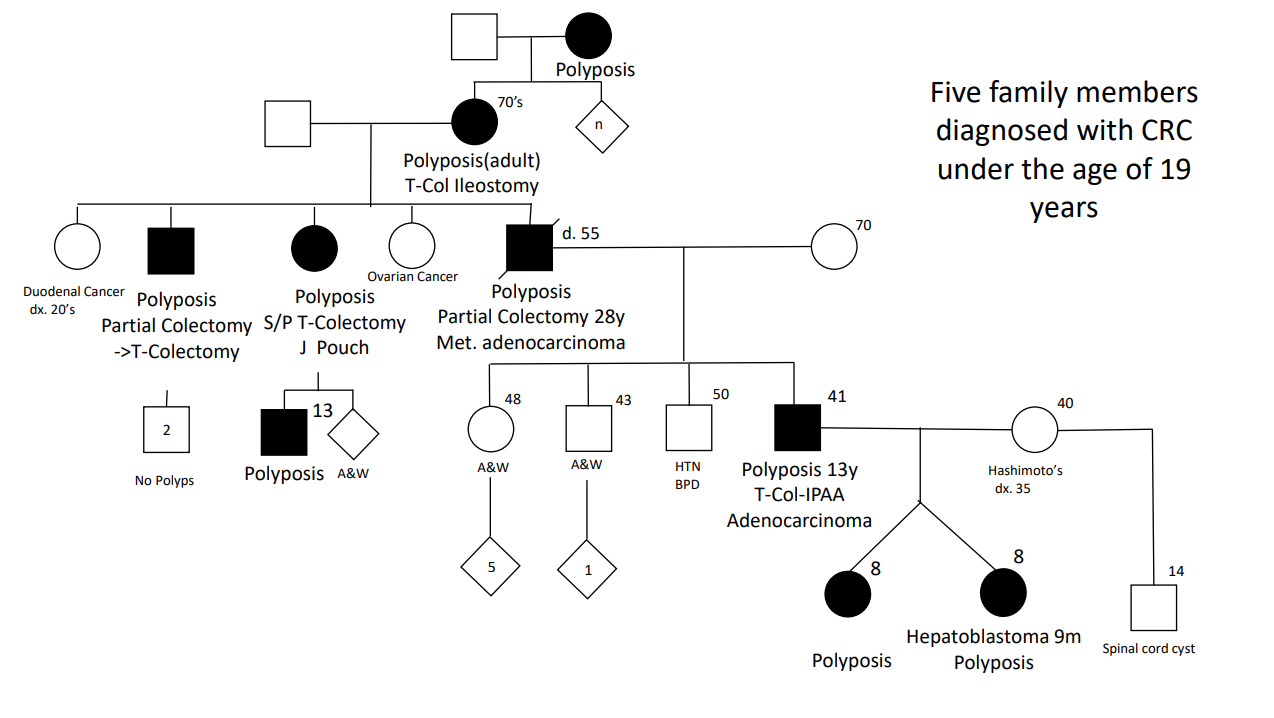
Case#2 presented 8 yo twins. Aggressive management was indicated as 5 family members developed colorectal cancer prior to age 20 years.
Case#2 Improvements in testing allowed identification of a point mutation in the 1B promoter region of the APC gene
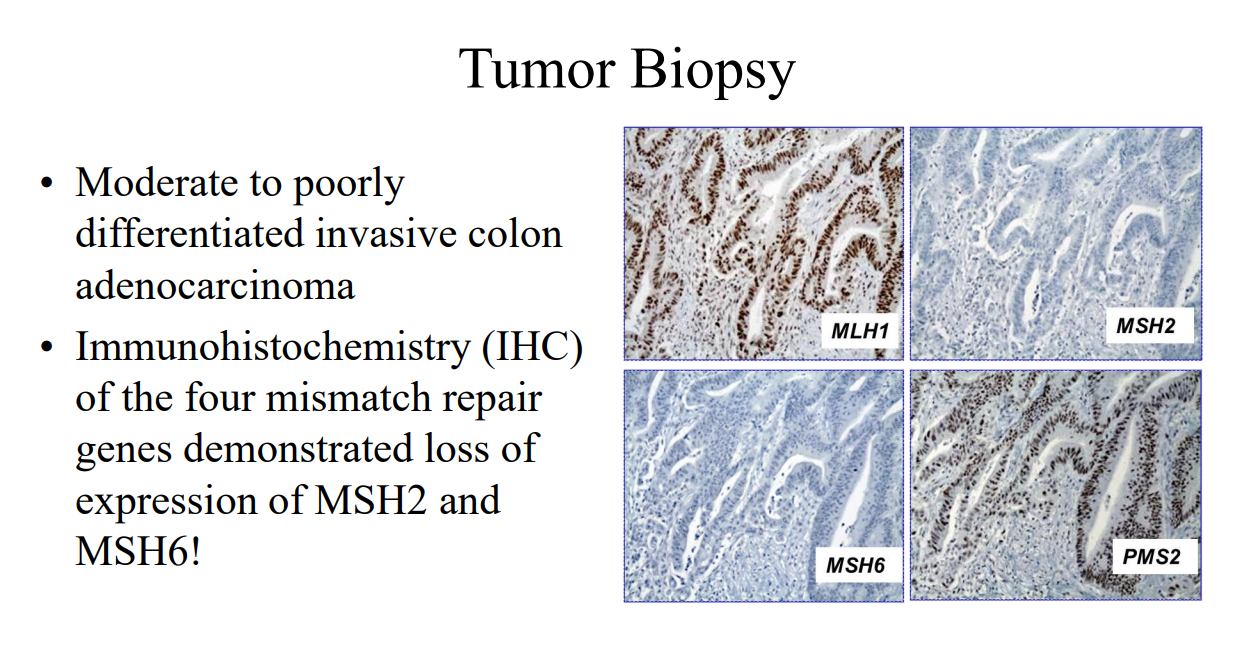
Case#3 presented a 16 yo with anemia and pain who was found to have a colonic mass related to mismatch repair mutation. Dr. Erdman indicated that obtaining adequate tissue for a diagnosis (“dig a hole”) is important. (As an aside, other colleagues have had the experience of tumors which were highly vascular and it is important to keep this possibility in mind)
Amsterdam II Criteria for Lynch Syndrome
Case#4 presented a 12 yo with neurofibromatosis (NF-1) who developed CRC and ultimately diagnosed with CMMR-D. This is a highly aggressive cancer susceptibility disorder with a very poor prognosis (see post: Are you familiar with CMMR-D?)
Case#5 presented two siblings (13 yo, 17 yo) who had half-sibling who died from CRC at age 25 yrs. This case illustrated “genetic anticipation” as each generation in this family with Lynch syndrome tended to develop CRC earlier in life. Amsterdam criteria can be helpful in identifying Lynch syndrome
Disclaimer: This blog, gutsandgrowth, assumes no responsibility for any use or operation of any method, product, instruction, concept or idea contained in the material herein or for any injury or damage to persons or property (whether products liability, negligence or otherwise) resulting from such use or operation. These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. Because of rapid advances in the medical sciences, the gutsandgrowth blog cautions that independent verification should be made of diagnosis and drug dosages. The reader is solely responsible for the conduct of any suggested test or procedure. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
Recommendation 5: Despite the presence of gastric polyps in children, and the later risk of duodenal polyposis and ampullary cancer in adult practice, there is no justification to commence routine UGI surveillance until the age of 25 years. (weak recommendation, low-quality evidence, consensus agreement 90%)
Methods: A prospective record of all upper GI endoscopies in children (aged 9 to 17) with FAP was kept across a 12-year period
Key finding:
Thirty-eight esophagogastroduodenoscopies (79%) identified at least 1 gastric or duodenal polyp in 22 (79%) patients; 10 (36%) patients had gastric adenomas
Eight (29%) patients showed very high numbers of polyps
All 21 patients who had duodenal polyps had adenomas
No patients had malignancy
My take: This is a provocative study. Is there a benefit for the patient in identifying gastric/duodenal polyps at a younger age?
A recent retrospective study (S Bonilla et al. JPGN 2020; 71: 288-291.Long-term Use of Bisacodyl in Pediatric Functional Constipation Refractory to Conventional Therapy) provides some reassuring information about the use of bisacodyl for pediatric constipation, n=164. Bisacodyl’s mechanism of action is due to its ability to cause mucosal secretion and a prokinetic effect on colonic mucosa.
Key findings:
Bisacodyl median dose was 5 mg/day, median duration of treatment was 14 months
Median number of BM/wk doubled after initiation of bisacodyl from 2 to 4 bm/w (P < 0.001)
Approximately 57% of patients had successful response. At long-term follow-up 55% of patients were successfully weaned off bisacodyl (median time of 18 months)
Side effects: 8 patients reported abdominal pain, 4 had diarrhea, and 1 had nausea
Limitations: open-label study, retrospective study, lack of a placebo-control
My take (from authors): “We observed no long-term complications with its long-term use in children.” Prospective studies are needed.
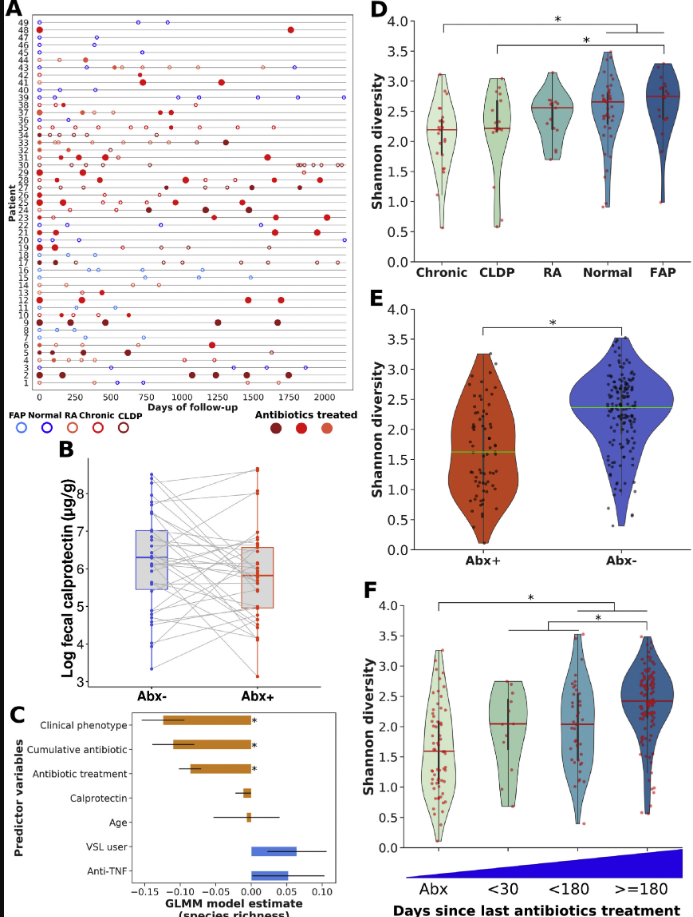
A prospective study (V Dubinsky et al. Gastroenterol 2020; 158: 610-24) followed 49 patients who had undergone pouch surgery for ulcerative colitis or for familial adenomatous polyposis (FAP).
The authors followed multiple parameters including calprotectin, metagenomes/bacterial diversity, antibiotic resistance testing, and virulence factors/toxins. 33 patients received antibiotics for a median of 425 days. Most patients were treated with a combination of ciprofloxacin and metronidazole.
Pouch phenotype: normal from UC (n=10), recurrent acute pouchitis (n=6), chronic pouchitis and Crohn’s-like disease of the pouch (n=27), and normal from FAP (n=6)
79% of antibiotic-treated patients had a clinical response to each course of antibiotics
89% of those who completed a 4-week course relapsed within 3 months
Median calprotectin values decreased by 40% in response to antibiotics
Antibiotic treatment reduced disease-associated bacteria including Clostridium perfringens, Ruminococcus gnavus, and Klebsiella pnneumoniae. However, F prausnitzii, a putative anti-inflammatory species, also decreased during antibiotic treatment
While antibiotic resistance was noted, these strains had a tendency toward lower potential for virulence and “did not induce secretion of inflammatory cytokines by epithelial cells”
Why do patients become antibiotic-dependent?
“We observed a drastic shift in microbiome composition on antibiotics cessation, characterized by blooms of nonintestinal bacteria, especially those originating from the oral cavity, as well as of opportunistic pathogens. Intestinal colonization by oral bacteria has been associated with UC and Crohn’s disease, and shown to trigger severe intestinal inflammation in germ-free mice…[this] drug-resistant microbiome may be fragile and unable to prevent colonization by exogenous bacteria that are ecologically fitter once antibiotics are discontinued.”
My take: This study provides insight into how antibiotics improve pouchitis; namely, they reduce disease-associated bacteria and promote an antibiotic-resistant microbiome with lower inflammatory potential.
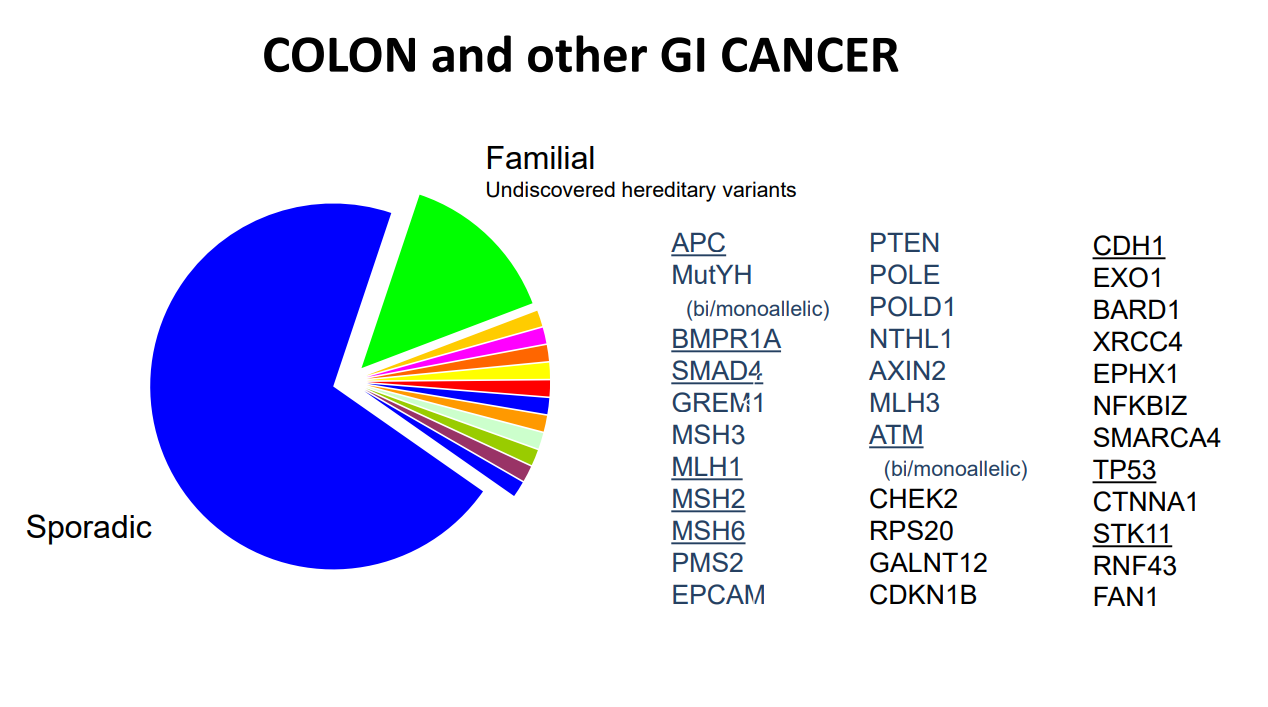
A recent cross-sectional study (PP Stanich et al. Clin Gastroenterol Hepatol 2019; 17: 2008-15, editorial 1942-44) identified a high frequency of genetic mutations among adults with at least 10 colonic polyps (cumulative burden of either adenomatous or hamartomatous).
This study had 3789 subjects who underwent multigene panel testing (MGPT) from 2012-16.
All subjects had at least 14 CRC-associated genes tested: APC, BMPR1A, CDH1, CHEK2, EPCAM, MLH1, MSH2, MSH6, MUTYH, PMS2, PTEN, SMAD4, STK11, TP53
A subset had 3 more newly recognized polyposis genes: GREM1, POLD1, and POLE
Key findings:
A mutation in at least 1 gene was found in 13.7%
In those with fewer than 20 cumulative adenomas, 7.6% had a disease-associated genetic mutation with the majority (5.3%) being nonpolyposis CRC genes
Younger patients, 18-29, were more likely to have mutations in any gene. For example, among patients with 10-19 polyps, these younger patients had a mutation in one of these genes in 27.8%; this is more than double the rate in any other age group.
Hamartomatous polyps, regardless of number, had a very high yield with genetic testing: 40% with 10-19 polyps and 72% with 20-99 polyps.
Limitations:
There is a referral bias in that the population was derived from a testing laboratory (Ambry)
In clinical practice, genetic testing frequently results in variants of unknown significance
My take: This study shows that genetic mutations are fairly frequent in patients with cumulative polyp burden of 10 or more, especially in younger age groups. The surprising finding is the high frequency of nonpolyposis CRC genes. Thus, in patients with adenomatous polyposis, testing beyond APC and MUTYH may be needed.
ESPGHAN Juvenile Polyposis Syndrome Recommendations These recommendations are different in that they do not recommend EGD in the pediatric age group: “Surveillance of the upper GI tract in affected or at-risk JPS patients is not required in childhood or teenage years, unless there is unexplained anaemia or upper GI symptoms.”
Are You Familiar with CMMR-D? The term CMMR-D refers to constitutional mismatch repair deficiency. This occurs when an individual inherits two MMR gene defects (rather than one gene defect in Lynch syndrome); with CMMR-D screening recommendations include yearly endoscopic evaluation beginning at age 3 years or at diagnosis.
A recent review article (SP MacFarland et al. JPGN 2019; 69: 273-80) provides clearcut guidelines on polyposis syndromes in pediatric patients.
Table 1 lists the syndrome, the mutated gene (s), and recommended screening (onset & interval). The article and table provide more nuance/guidance but the basic recommendations are noted as follows:
For Familial Adenomatous Polyposis (FAP), the authors recommend onset of colonoscopy at 10 years and with 1 year intervals. “Colectomy recommended by 20 to 25 years.” EGD is recommended at 18 to 20 years. Thyroid ultrasound is recommended at 18 years. Alpha-fetoprotein levels to check for hepatoblastoma are recommneded every 3-6 months in infancy up to 5 years of age.
For Juvenile Polyposis Syndrome, EGD and Colonoscopy are recommended at 15 years with interval evaluations at 1-3 years.
For Peutz-Jeghers syndrome, EGD and Colonoscopy are recommended at 8 to 10 years (along with small bowel evaluation with either MRE or video capsule). Interval followup is recommended every 2-3 years.
Table 2 provides suggestions for familial screening in pediatric polyposis syndromes.
ESPGHAN Juvenile Polyposis Syndrome Recommendations These recommendations are different in that they do not recommend EGD in the pediatric age group: “Surveillance of the upper GI tract in affected or at-risk JPS patients is not required in childhood or teenage years, unless there is unexplained anaemia or upper GI symptoms.”
Are You Familiar with CMMR-D? The term CMMR-D refers to constitutional mismatch repair deficiency. This occurs when an individual inherits two MMR gene defects (rather than one gene defect in Lynch syndrome); with CMMR-D screening recommendations include yearly endoscopic evaluation beginning at age 3 years or at diagnosis.
Management of Familial Adenomatous Polyposis in Children and Adolescents: A Position Paper from the ESPGHAN Polyposis Working Group: Link: JPGN 2019; 68 (3): 442-441
Unlike some working groups, the working group for ESPGAN Polyposis did not equivocate in making clear cut recommendations, especially for recommendations #6a and #6b, for Familial Adenomatous Polyposis (FAP) in children.
SOME OF THE RECOMMENDATIONS Recommendation 1: Predictive genetic testing should be offered to at-risk children at the age of 12 to 14 years. Families should receive genetic counselling before and at the time of testing. Children who are symptomatic with rectal bleeding should undergo earlier testing. (weak recommendation, low-quality evidence, consensus agreement 100%)
Recommendation 3: In those confirmed to have FAP on predictive genetic testing, and those considered at risk where genetic testing is not possible, colonic surveillance should commence age 12 to 14 years. Once adenomas have been identified, intervals between surveillance colonoscopy should be individualized depending on colonic phenotype every 1 to 3 years. Rectal bleeding or mucous discharge should lead to a colonoscopy at any age. (weak recommendation, low-quality evidence, consensus agreement 100%)
Recommendation 4: Colectomy is necessary to prevent CRC in adulthood. Decision on the timing for colectomy should be determined by polyp burden and characteristics of colonic adenomas in the context of social, personal, and educational factors. IRA or IPAA have their merits and disadvantages and many factors impact on the choice of surgery. The choice should be based on patient phenotype (rectal and colonic burden) and genotype, at the discretion of the surgeon. (weak recommendation, low-quality evidence, consensus agreement 100%)
Recommendation 5: Despite the presence of gastric polyps in children, and the
later risk of duodenal polyposis and ampullary cancer in adult practice, there is no justification to commence routine UGI surveillance until the age of 25 years.
(weak recommendation, low-quality evidence, consensus agreement 90%)
Recommendation 6a: Routine screening for HPB [Hepatoblastoma] in patients with FAP is not recommended. In children found to have HPB, there is no evidence that routine genetic testing or endoscopic screening for FAP is required.
(weak recommendation, low-quality evidence, consensus agreement 100%)
Recommendation 6b: Children with bilateral and multiple CHRPE [congenital hypertrophy of retinal pigment epithelium] lesions should undergo colonoscopy at age 12 to 14 years. If CHRPE lesions are single or unilateral in the absence of relevant family history, further evaluation should not be required. (weak recommendation, low-quality evidence, consensus agreement 100%)
Recommendation 6c: The vast majority of desmoids tumours are sporadic; children identified to have a DT have approximately 10% risk of FAP. If the kindred is known to have FAP and the child has a desmoid, it should be presumed the child has FAP. In a child presenting with a DT, testing the DT for a b-catenin /CTNNB1 mutation is recommended. If a b-catenin /CTNNB1 mutation is found, this indicates sporadic desmoid and further investigations for FAP are not required. If b-catenin /CTNNB1 mutation is not found, the patient should be investigated for FAP. (weak recommendation, low-quality evidence, consensus agreement 100%)
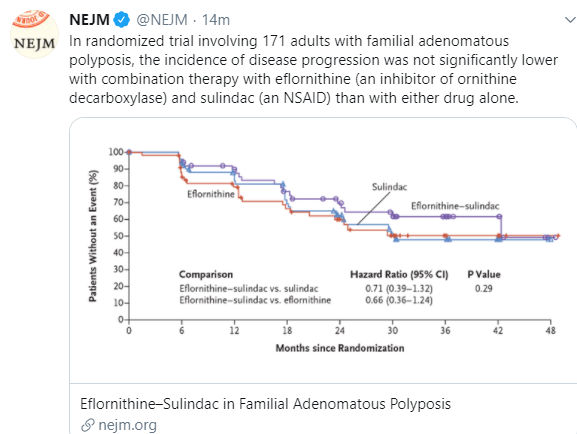
Recommendation 7: There is no role for the use of chemoprevention agents in
children with FAP. (strong recommendation; moderate-quality evidence, 100%
consensus)
Disclaimer: These blog posts are for educational purposes only. Specific dosing of medications (along with potential adverse effects) should be confirmed by prescribing physician. This content is not a substitute for medical advice, diagnosis or treatment provided by a qualified healthcare provider. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a condition.
In their introduction (KP Quinn et al. Clin Gastroenterol Hepatol 2016; 14: 1296-1301), the authors state the following: “Despite the widely held notion that pouchitis is a rare complication in FAP following IPAA, clinical experience at our institution suggests [it]…is underestimated.”
Methods: retrospective cohort study of all FAP patients who underwent IPAA (ileal ouch-anal anastomosis) from 1992-2015 at their institution (Mayo clinic), n=113.
Key findings:
25 (22.1%) developed pouchitis with a mean time to pouchitis of 4.1 years.
Of the 25 who developed pouchitis, 72% had an acute course and 28% had a chronic course.
My take: While pouchitis does occur more commonly in IBD following IPAA, it does occur with FAP more frequently than previously described.
Here’s a link to abstract: Updated Guidelines on Genetic Testing/Management for Hereditary GI Cancer Syndromes (The American Journal of Gastroenterology110, 223-262 (February 2015) | doi:10.1038/ajg.2014.435). This ACG guideline specifically discusses genetic testing and management of Lynch syndrome, familial adenomatous polyposis (FAP), attenuated familial adenomatous polyposis (AFAP), MUTYH-associated polyposis (MAP), Peutz–Jeghers syndrome, juvenile polyposis syndrome, Cowden syndrome, serrated (hyperplastic) polyposis syndrome, hereditary pancreatic cancer, and hereditary gastric cancer.
I glanced at the guideline –it is about 40 pages in length. It provides a lot of in-depth information on these infrequent disorders.