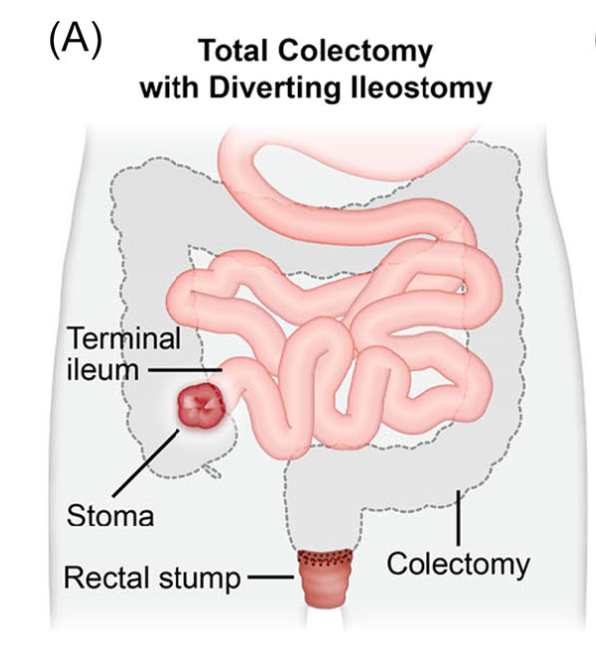
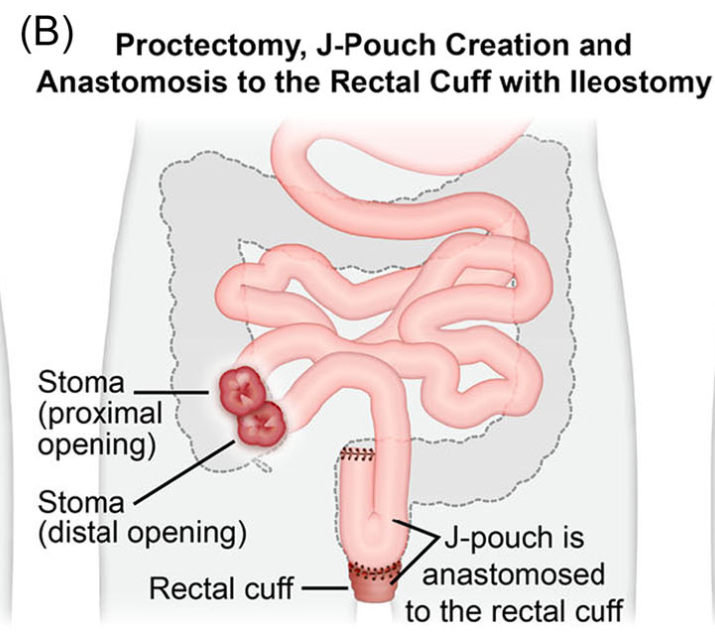
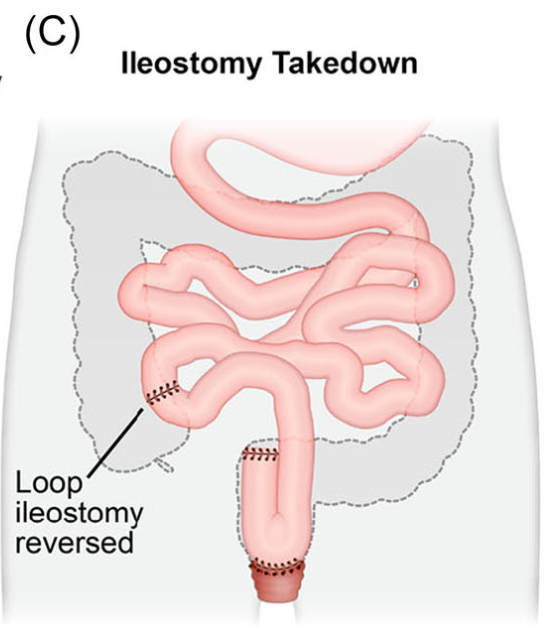
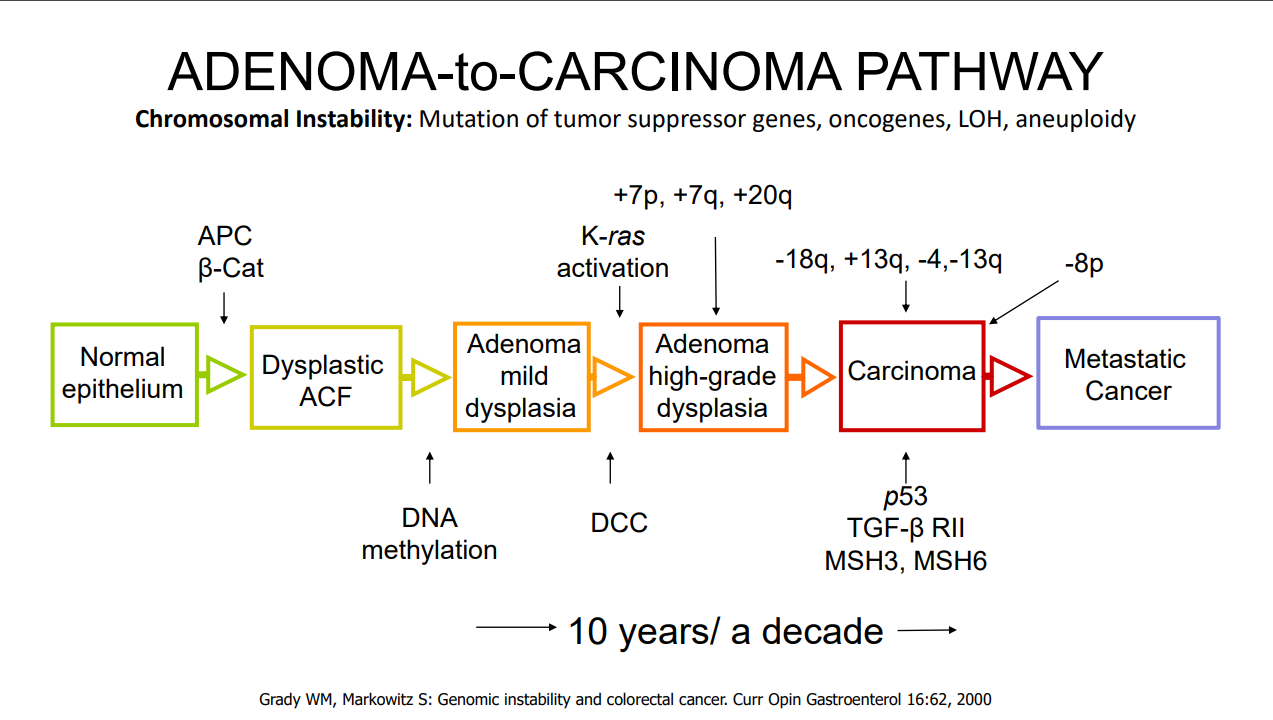
- H Bouchiba et al. Gastroenterol 2026; 170: 942-952. Open Access! Incidence of cancer in patients with familial adenomatous polyposis in the Netherlands: a nationwide cohort study
- J Karstensen. Gastroenterology, 2026; 170, 884-886 (editorial). Open Access! From Old Battles Won to New Frontiers in Familial Adenomatous Polyposis
- D Terlouw et al. Gastroenterol 2026; 170: 932-941. Open Access! Prevalence and Consequences of APC Mosaicism in Patients With Colorectal Adenomas
From Bouchiba et al:
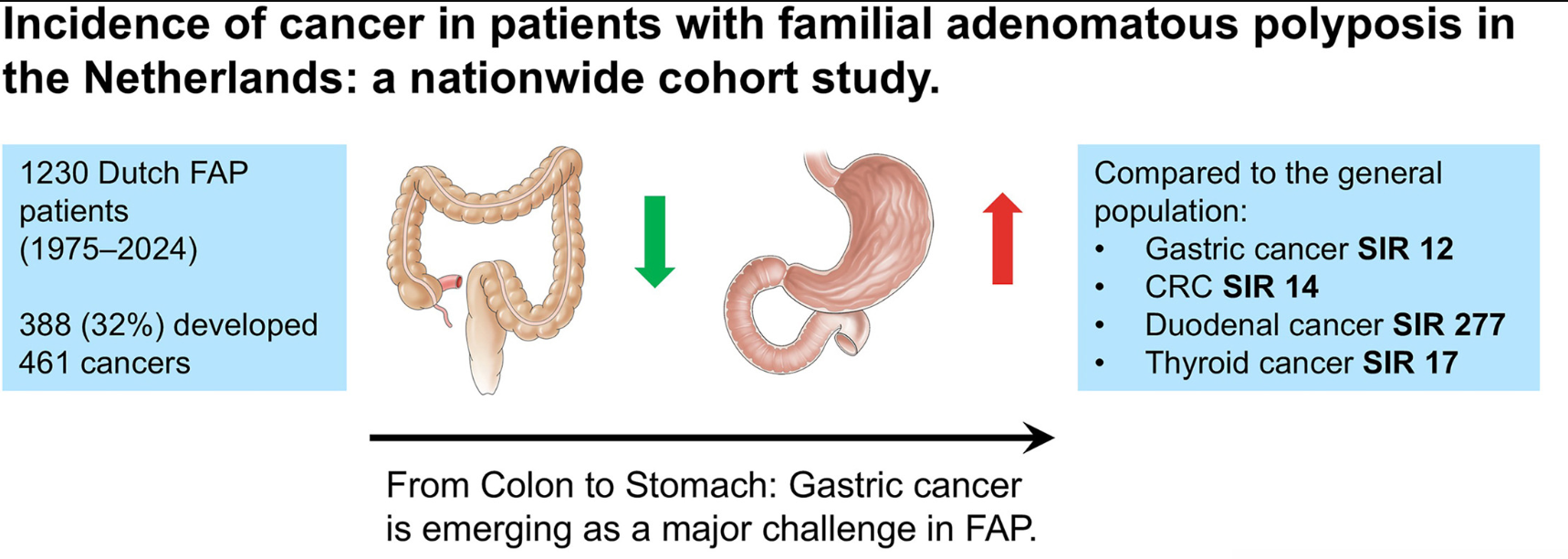
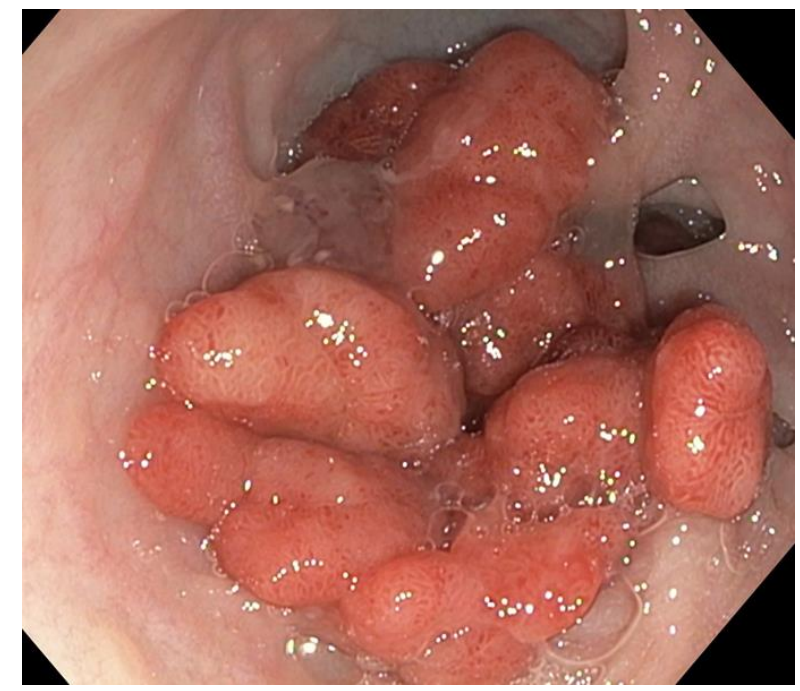
From the editorial: “Bouchiba et al10 present a Dutch cohort study providing the most comprehensive evaluation of cancer incidence and temporal trends in FAP to date. Among 1230 patients with FAP followed over 5 decades, one-third developed cancer despite major preventive advances, underscoring the persistent lifetime risk in FAP…the 12-fold higher risk compared with the general population and the frequent late-stage detection of gastric cancer are concerning and underscore the need for improved endoscopic surveillance strategies.”
From Terlouw et al:
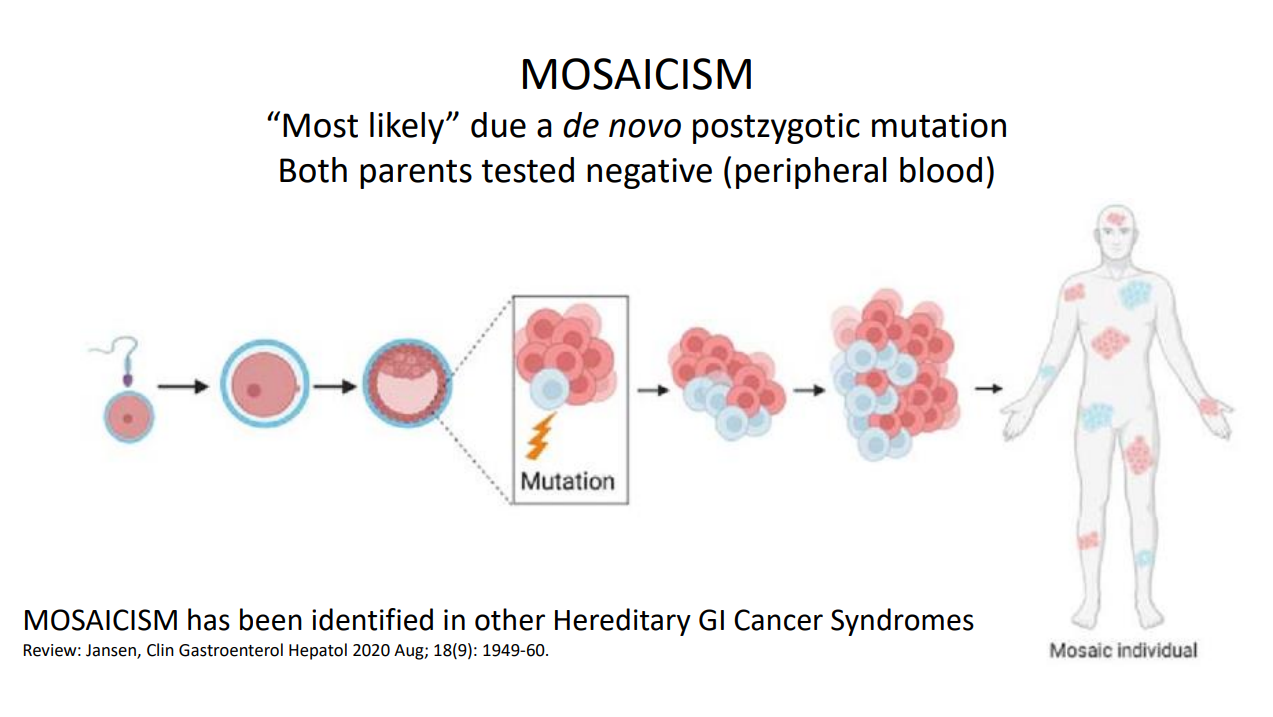
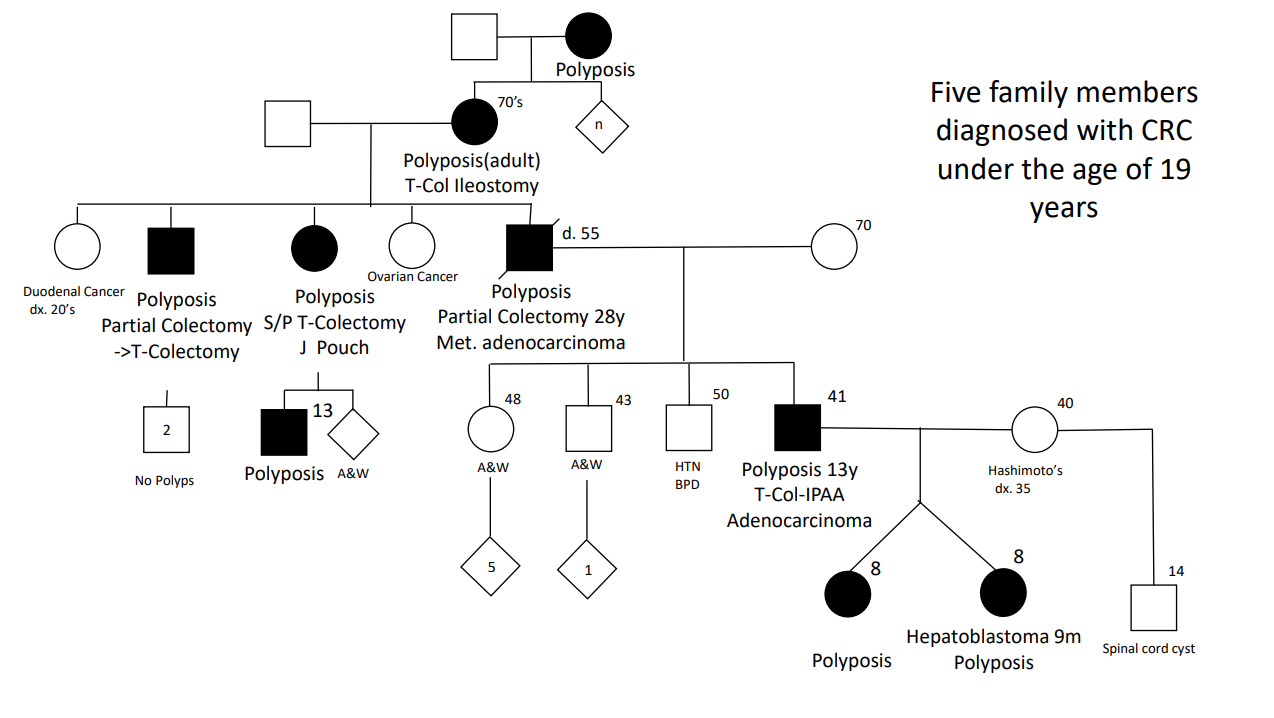
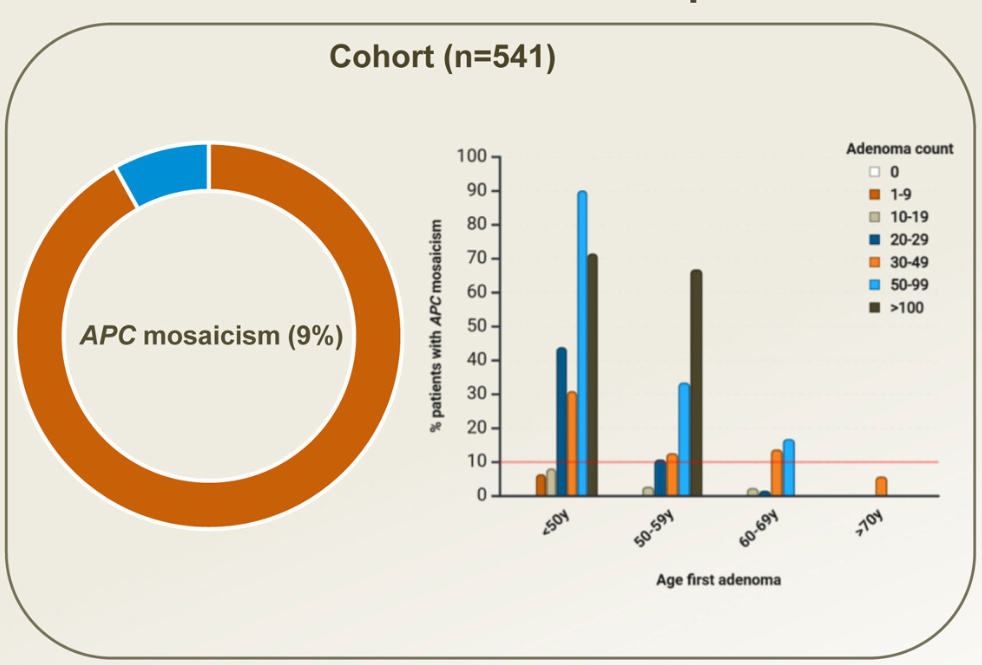
Background: A substantial proportion of patients with adenomatous polyposis have no germline pathogenic variant in APC. The aim of this study was to determine the prevalence of APC mosaicism in these patients with unexplained polyposis
Key findings:
- The rate of APC mosaicism was 9.4%. This rate was 14.3% (46 of 322) in patients who met the scope of national hereditary polyposis testing guidelines (≥10 adenomas before the age of 60 or with ≥20 adenomas before the age of 70)
- In patients who did not meet the scope of national guidelines, the detection rate was 2.3% (5 of 219)
- None of the children tested in this cohort inherited the mosaic variant.
My take:
- Despite increasing knowledge of FAP, frequent cancers continue to occur
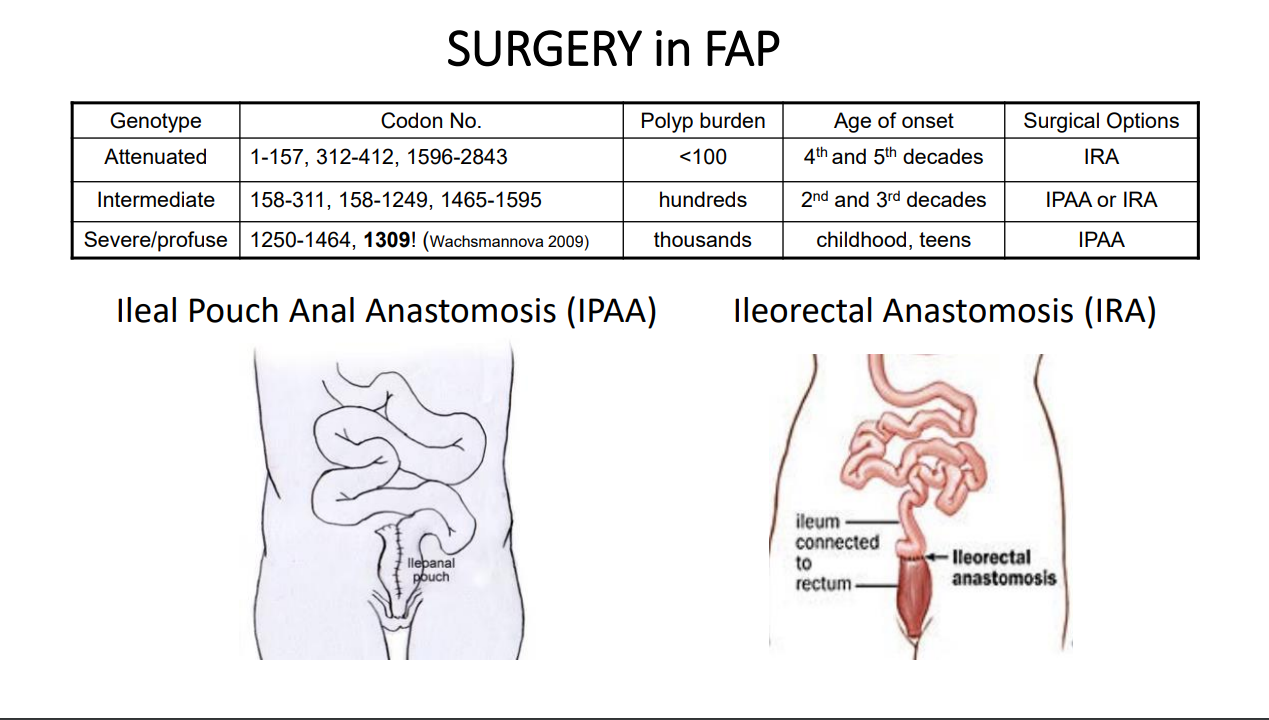
- In those with features of FAP without identified pathogenic mutations, testing for APC mosaicism should be considered. In addition, in those with APC gene mutation, regular colonoscopy is recommended along with at least one esophagogastroduodenoscopy.
Related blog posts:
- Genetic Risk Assessment and Testing for Gastrointestinal Cancers and Polyposis (2025)
- Management of Colorectal Adenomas in Adolescents
- When Are UGI Polyps Important for Familial Adenomatous Polyposis?
- What I Like About ESPGHAN Familial Adenomatous Polyposis Position Paper (2019)
- Dr. Steve Erdman: Perplexing Polyposis Patients: a Case-Based Discussion
- Approach to Fundic Gland Polyps and VCE for Polyposis Syndromes
- Gastric Polyposis in 16 Year-Old