In June 2024 (special issue), Gastroenterology published an entire issue (193 pages) focused on celiac disease. There was a lot of useful information on almost every aspect of this disease. Below I have summarized some of the points –this is the last of 4 posts on this issue.
In addition to these articles, another good update on aspects of CeD was the Bowel Sounds podcast with Arun Singh. Link: Clinical Conundrums in Celiac Disease.
DW Adams et al. Gastroenterol 2024; 167: 51-63. Clinical Presentation and Spectrum of Gluten Symptomatology in Celiac Disease
This article highlights the myriad presentations of CeD. Due to screening of at-risk groups. fewer patients are presenting with malabsorption and many patients at diagnosis are overweight or obese. In adults, 15-28% are overweight and 7-11% are obese.
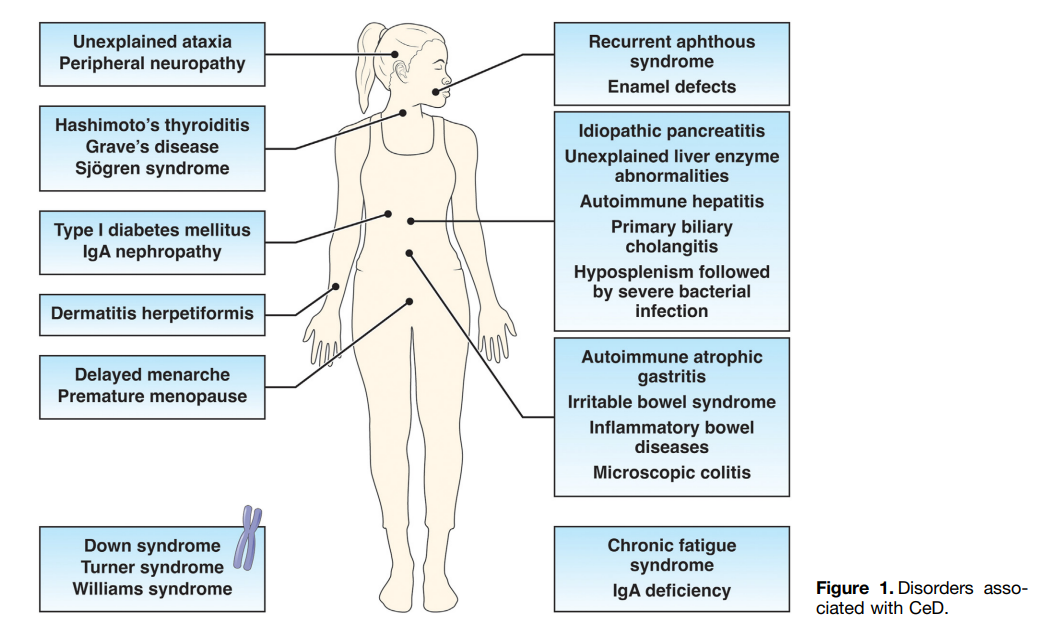
- Hematology: Among adults with iron-deficiency anemia, 1 in 31 have histologic evidence of CeD
- Skin: Dermatitis herpetiformis
- Neurologic: Headache, brain fog, peripheral neuropathy, ataxia
- Oral: dental enamel defects, aphthous stomatitis
- Musculoskeletal/rheumatologic: osteopenia/osteoporosis/fractures, arthralgias/arthritis. Low bone mass with “z-scores less than -2 SD for age is present in 6-16% of children” with CeD
- Growth in children: pubertal delay, short stature, growth failure
- Fertility: female infertility, spontaneous abortions, premature births, hypogonadism (both sexes)
- GI: stomach pain, diarrhea, bloating/distention, constipation, weight loss
- Liver: elevated liver enzymes in up to 40% of patients with CeD at diagnosis. 1 in 20 patients with cryptogenic cirrhosis have CeD. In children, ~25% have “modest elevations in liver enzymes” at diagnosis. “Normalization …is generally seen within 6-12 months of a GFD.”
- Endocrine: higher rates of type 1 diabetes and autoimmune thyroid disease
- Asymptomatic: “10-27% of CeD patients are asymptomatic at diagnosis…Asymptomatic patients with CeD were taller, had lower TTG-IgA levels, and had less severe intestinal damage.” However, “patients reporting to be asymptomatic often record improvement” with GFD.
The authors detail how gluten exposure triggers symptoms. Gluten ingestion activates gluten-specific T cells and acute increases in interleukin-2 (IL-2). IL-2 is postulated to cause neuro-enteric symptoms and leads to release of serotonin. Serum IL-2 is a biomarker for acute gluten exposure.
MI Pinto-Sanchez et al. Gastroenterol 2024; 167: 116-131. Nutrition Assessment and Management in Celiac Disease
- This article provides specific nutritional recommendations for patients with CeD including involvement of a dietitian. The authors note that some studies in children have found low iron, vitamin D, calcium, vitamin B12, vitamin B6, folate, zinc and magnesium in their GFD. They recommend checking for iron, ferritin, folate, vitamin B12, vitamin D, vitamin A, zinc, selenium, copper, and chromium at diagnosis.
- The adoption of a GFD “has been associated with an increased risk of MASLD likely related to the nutritional composition of packaged gluten-free foods.” (see: Gluten-Free Diet Can Be Unhealthy)
- 30% of CeD adult patients experience persistent symptoms. The most common cause is continued gluten exposure (often inadvertent). Gluten contamination elimination diet can be implemented in this setting “though not feasible as a long-term solution.” (See: What To Do For Pediatric Patients with Non-Responsive Celiac Disease)
My take: More recent studies have indicated that more limited nutrient testing is appropriate in the pediatric population (see: How to Provide More Cost-Effective Celiac Care and Nutrient Deficiencies with Celiac Disease). Many nutrient deficiencies (in pediatrics) improve with institution of GFD and other nutrient deficiencies are similar in frequency as the general population.
U Volta et al. Gastroenterol 2024; 167: 104-115. Diagnosis of Seronegative and Ultrashort Celiac Disease
- “Seronegative villous flattening is a very rare condition” in children…and is “often attributable to other diagnosis (eg. inflammatory bowel disease…). The authors provide a diagnostic algorithm. In this setting, first step is to check HLA-DQ2/DQ8. A negative test “virtually rules out CeD.” A positive test indicates CeD is possible if other conditions are excluded. This includes autoimmune enteropathy, immune deficiencies, medication effects, infections, and other conditions. (See:Seronegative Villous Atrophy and@AmyOxentenkoMD: Celiac Disease and Mimics and How Many Cases of Celiac Disease Are We Missing?)
- CeD confined to 1st portion of duodenum is considered ultra-short CeD. Algorithm for ultra-short CeD (Figure 2). The disease is supported by positive serology and positive HLA DQ2/DQ8. If serology and/or HLA DQ2/DQ8 are negative, it is very unlikely to be CeD and other diagnosis need to be identified (eg. peptic duodenitis).