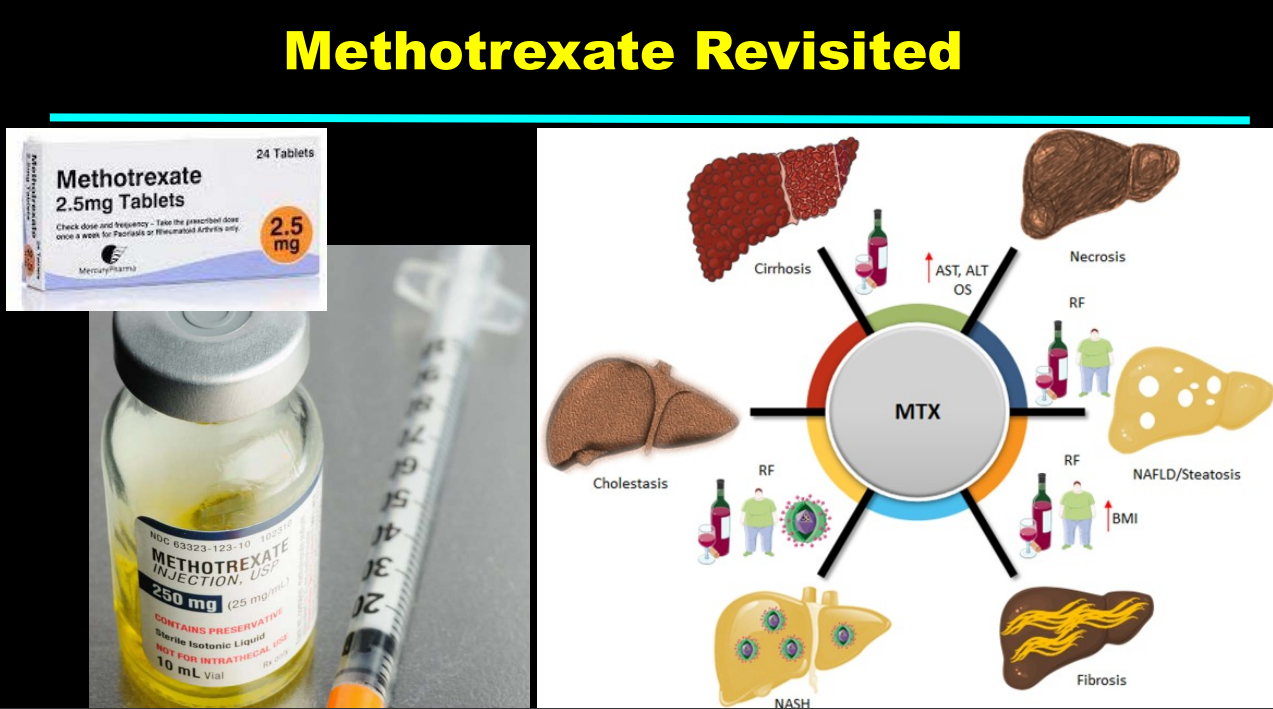
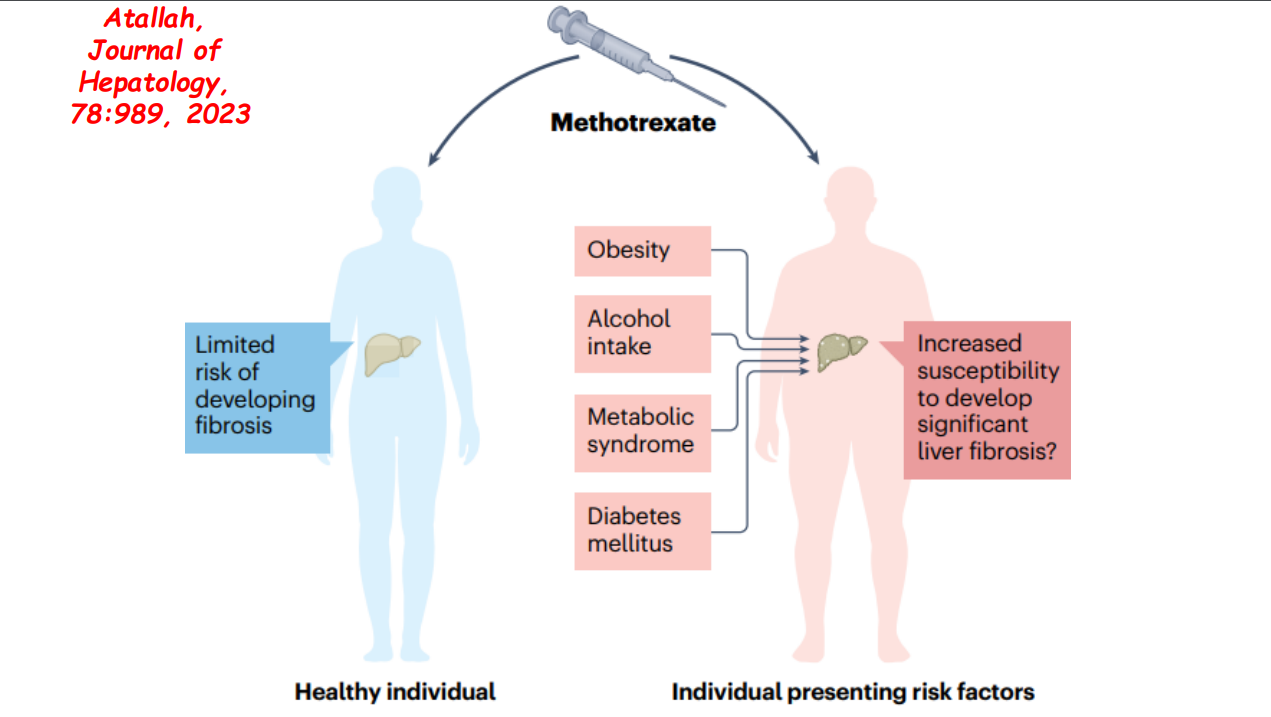
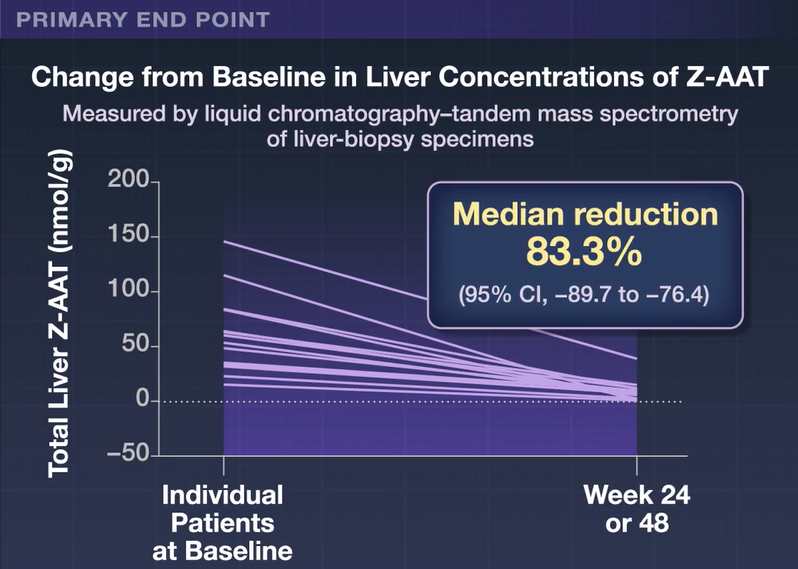
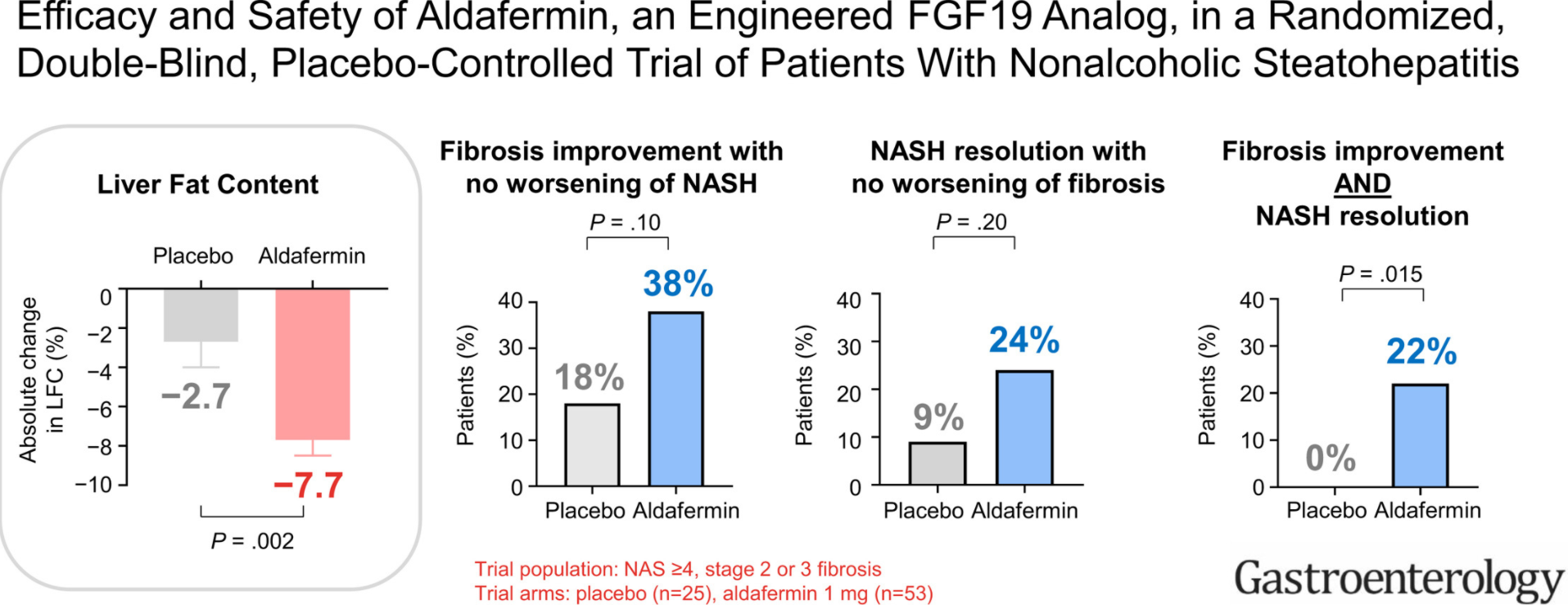
Recently Dr. Balistreri gave our group an excellent lecture. I have taken some notes and shared some slides. There may be inadvertent omissions and mistakes in my notes.
In my view, Dr.Balistreri’s contributions to our field of pediatric gastroenterology, hepatology and nutrition are unsurpassed by any other individual. This is due to his leadership roles (division director, president of AASLD and NASPGHAN), his editor roles (Journal of Pediatrics, and JPGN), his extensive publications/research including sentinel discoveries in bile acid pathophysiology and treatment, and through education (lectures and mentorship).

Key points:
- Fifty years ago, ~65% of neonatal cholestasis cases were poorly understood and lumped together under the heading of “Idiopathic Neonatal Hepatitis” (INH). The discovery of Alpha-One Antitrypsin (A1AT) Deficiency was instrumental, indicating that there were specific medical diseases mislabeled as INH
- A1AT deficiency proved that many cases were NOT “idiopathic,” NOT necessarily “neonatal,” and NOT “hepatitis”
- Currently, less than 10% of infants with neonatal cholestasis are unspecified. Dr. Balistreri’s goal has been to make sure every patient receives a precise diagnosis
- Now more than 90 genetic conditions have been recognized as causing neonatal cholestasis
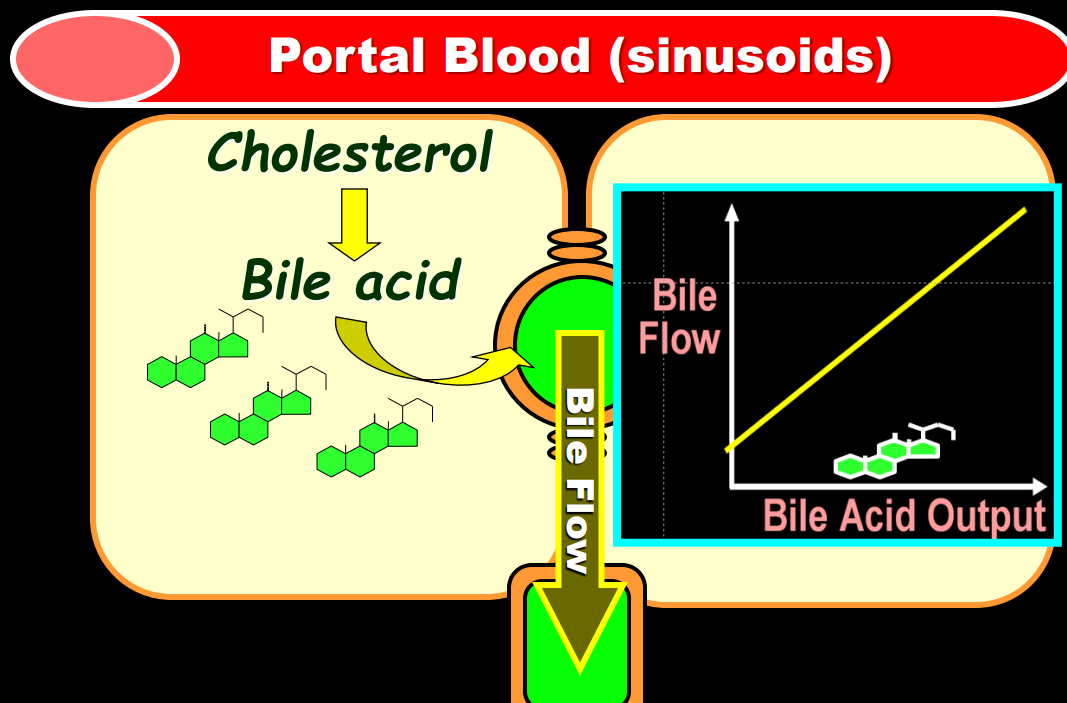
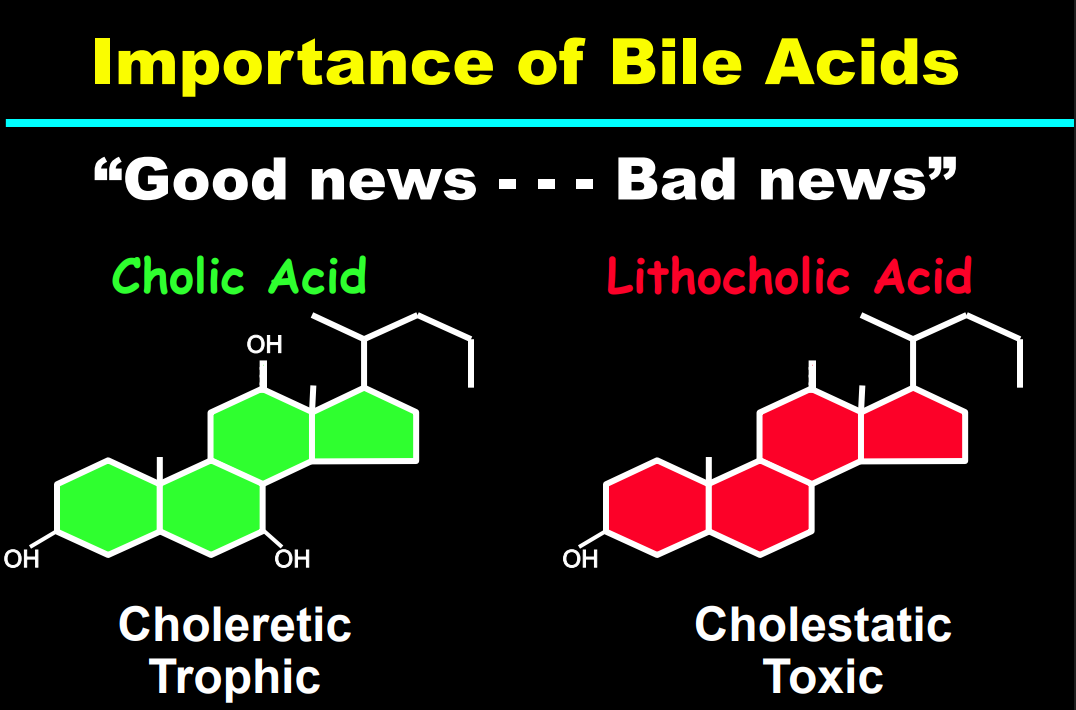
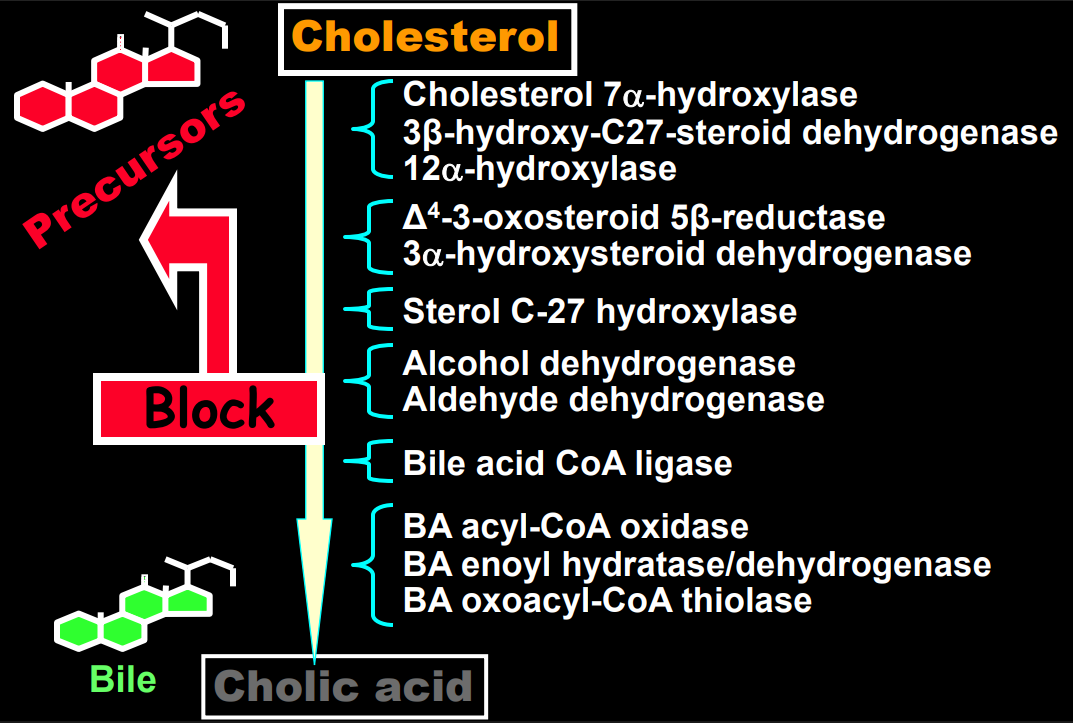
- The journey of unraveling the reasons for neonatal cholestasis includes the referral of two infants from Atlanta by Dr. Saripkin to Cincinnati. These infants who had an older sibling who had died at 4 months of age were determined to have an inborn error of bile acid metabolism. Subsequently, they were treated successfully with cholic acid which suppressed production of toxic precursors via feedback inhibition
- There are 11 steps in the conversion of cholesterol to bile acids; thus, it was hypothesized and later proven that there would be many other inborn errors of bile acid metabolism


















Related blog posts:
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 1)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 2)
- NASPGHAN22: History of Pediatric GI & Selected Slides from the William F Balistreri Lecture (Part 3)
- Good Review of Cholestatic Liver Diseases
- Aspen Webinar 2021 Part 8 -Neonatal Cholestasis (link to PreventionGenetics Panel)
- Year-in-Review for Pediatric Hepatology (2024)
- Online Aspen Webinar (Part 2) -Abnormal Liver Enzymes in a Tween (2022)
- Medical Progress: Toward Hepatitis C Elimination (2020)
- Immune Mediated Disorders Associated with TNF Inhibitors Can Involve the Liver Too
- Efficacy and Safety of Odevixibat with Alagille Syndrome (ASSERT Trial)
- Relooking at 6-Year Data of Maralixibat for Alagille Syndrome
- Six Year Data for IBAT Inhibitor Treatment for Alagille Syndrome
- Lecture: IBAT Inhibitor for Alagille Syndrome
- GALA: Alagille Study
- NASPGHAN Alagille Syndrome Webinar
- Intracranial Hypertension & Papilledema with Alagille Syndrome
- Explaining Differences in Disease Severity for Alagille Syndrome
- Gene Therapy for Alpha-One Antitrypsin Deficiency
- Alpha-1-Antitrypsin Deficiency
- Identifying Biliary Atresia in Infants: New Guidelines
- Updated Diagnostic Accuracy of Serum Matrix Metalloproteinase-7 (MMP-7) for Biliary Atresia
- Why Didn’t Screening for Biliary Atresia Improve Outcome In This Study?